Originally posted by Klute
Well, it could be worth a try. I am a little sceptical I must admit, although copper metal will surely be formed, it might not have the catalytic
properties the calcined, H2-reduced catalyst has. I don't think you can reduce it in aqueous conditions and still expect copper hydride to form.
|


















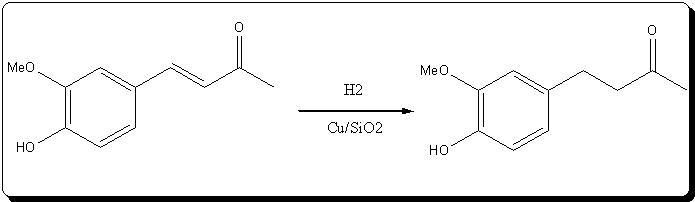










 ) or you're using a DEhydrogenation catalyst for hydrogenation?
) or you're using a DEhydrogenation catalyst for hydrogenation? I'd much rather use Zn/H+ any day...
I'd much rather use Zn/H+ any day...