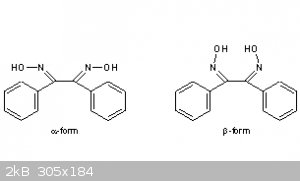
Literature References: Three isomers occur: a or anti, b or syn, g or amphi. Prepn of a-form from benzil and hydroxylamine hydrochloride: Brady,
Perry, J. Chem. Soc. 127, 2874 (1925); F. J. Welcher, Organic Analytical Reagents vol. III (Van Nostrand, New York, 1947) pp 224-227; Boyer et al., J.
Am. Chem. Soc. 77, 5688 (1955). Prepn of b-form: Brady, Perry, loc. cit.; Boyer et al., loc. cit. Prepn of g-form: Welcher, loc. cit., pp 227-228; see
also Boyer et al., loc. cit.
Derivative Type: a-Form
Properties: Crystals from methanol, mp 238-240°, Boyer et al., loc. cit., also reported as mp 243-244°, Meisenheimer, Lamparter, Ber. 57, 276
(1924). Practically insol in water, ether, glacial acetic acid; slightly sol in alc; readily sol in NaOH solns.
Melting point: mp 238-240°; mp 243-244°, Meisenheimer, Lamparter, Ber. 57, 276 (1924)
Derivative Type: b-Form
Properties: Crystals, mp 212-214°, Meisenheimer, Lamparter, loc. cit.
Melting point: mp 212-214°, Meisenheimer, Lamparter, loc. cit |



