
Bedlasky - 3-10-2022 at 19:52
Hi!
Several months ago I played a lot with bromatometric titrations. I find them quite useful. So today I post instructions for determination of phenols
and anilines. O-alkylphenols require different conditions, so I describe procedure for them separately. I hope that some organic chemist find this
useful.
This type of reaction without catalyst is possible due to strong electron-donating capabilities of -OH, -NH2 and -OR groups.
Chemicals used
0,1M KBrO3 (this needs to be exact concentration, mine was 0,1000 mol/l)
0,1M Na2S2O3 (again, exact concentration, mine was 0,0996 mol/l)
0,55M KBr
KI
1+1 HCl
Glacial acetic acid
Salicylic acid
Aniline
Anisole
1M NaOH
Advantage of KBrO3 is that it is primary standard, so no need for standardization. Sodium thiosulfate can be standardized using potassium dichromate,
bromate or iodate. More about this here.
Determination of phenols and anilines
BrO3- + 5Br- + 6H+ --> 3Br2 + 3H2O

Br2 + 2I- --> 2Br- + I2
I2 + 2S2O32- --> 2I- + S4O62-
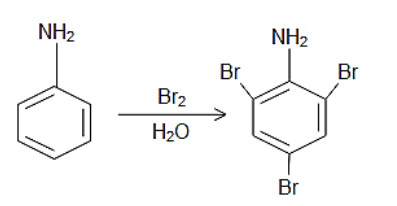
cca 0,47 g of aniline (weighed to 4 decimal places) is dissolved in beaker containing water and 5 ml 1+1 HCl. Content of the beaker is quantitatively
transferred in to the 100 ml volumetric flask and flask is filled to the mark.
In to the beaker with cca 0,69 g of salicylic acid (weighed to 4 decimal places) and some water is added 1M NaOH dropwise until all salicylic acid is
dissolved. Solutions is quantitatively transferred into the 100 ml volumetric flask and flask is filled to the mark.
Procedure for aniline/salicylic acid is the same from now on.
2x10 ml aliquot from volumetric flask is pipette out (using bulb pipette) to two ground glass Erlenmeyer flasks. Then 2x5 ml of 0,1M KBrO3 (using bulb
pipette) and 2x20 ml of 1+1 HCl are added. Finally 2x5 ml of 0,55M KBr is added, walls of flasks are quickly washed with distilled water and flasks
are immediately stoppered with ground glass stoppers (to prevent loss of bromine) and put in to the dark.
Reaction mixtures are then let stand for 20 minutes. After that 2x0,8 g of solid KI is added to Erlenmayer flasks, flasks are again stoppered and put
in to the dark. They are left for another 10 minutes. Liberated iodine is then titrated with 0,1M Na2S2O3. Before titration 2x5 ml of DCM is added to
dissolve tribromoaniline/phenol. When solution become just light yellow, add some starch to finish titration more precisely. Or you can use DCM as
sort of indicator and end titration when DCM layer become colourless.
This method can be used for determination of other C-alkylated phenols/anilines. Electron-donating groups on aromatic ring shouldn't interfere with
reaction. -COOH and -SO3H groups are removed from the ring during reaction as CO2 and H2SO4. But I am not sure if strong electron-withdrawing groups
(like -NO2, -CN etc.) don't spoil the reaction. I have some nitrophenol, so I can try in the future.
Bromination of O-alkyphenols is incomplete at these conditions. O-alkylphenols can be determined according to the method below. N-alkylanilines can't
be determined using this method.
Determination of O-alkylphenols
BrO3- + 5Br- + 6H+ --> 3Br2 + 3H2O
Br2 + 2I- --> 2Br- + I2
I2 + 2S2O32- --> 2I- + S4O62-
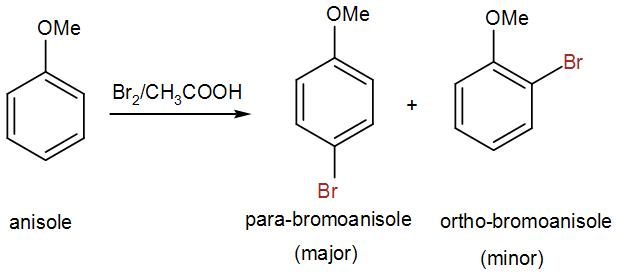
2x0,16 g (weighed to 4 decimal places) of anisole is added to the Erlenmayer flasks containing 2x5 ml of 0,1M KBrO3 and 2x5 ml of 0,55M KBr. 2x90 ml
of glacial acetic acid is then added and flasks are immediately stoppered and put in to the dark. After 20 minutes 2x90 ml of water and 0,8 g of solid
KI are added. Flasks are stoppered and put in to the dark. After 10 minutes liberated iodine is titrated with 0,1M Na2S2O3. When solution become just
light yellow, add some starch to finish titration more precisely.
I tried the same procedure for N-ethylaniline and N,N-diethylaniline, but after finishing titration with thiosulfate, some iodine is slowly liberated
from the reaction mixture. These N-alkylanilines are probably oxidized to some product, which slowly release bromine, which oxidize iodide to iodine.
So this procedure can't be applied to N-alkylanilines.
[Edited on 4-10-2022 by Bedlasky]
[Edited on 11-20-2023 by Texium]
Fery - 4-10-2022 at 09:58
Bedlasky nice experiments !!!
I'm not good in theory but couldn't the substituted anilines behave like bases and cause disproportionation of bromine into hypobromate which could
further change into bromate? I do not know whether they are more basic than aniline? But you used excess of HCl so the environment was certainly
acidic, wasn't it?
I'm also happy that you found an usage for the anisole.
If you need nitrophenol I can send you orto as well para (commercial samples) though I'm not sure whether they couldn't oxidize to quinones?
Bedlasky - 4-10-2022 at 12:48
Thanks Fery! Reaction is carried in huge excess of HCl, so disproportionation can't happen.
Pumukli - 8-10-2022 at 04:36
Thank You Bedlasky for your work in organic analysis!
This is a neglected part of chemistry here on the Board. (What if we had a dedicated "thread" or "sub-forum" or "section" or something like that on
analytical methods? I think it would not become the most deserted section. :-) )
Being able to determine some chemical species quantitatively is a "higher quality" amateur chemistry in my eyes. ;-)
Regarding your work I think that the determination of phenols/phenol ethers and anilines is important. They are so widespread around us, relatively
easy to acquire and can be subjected to numerous transformations. Bromatometry is also a versatile method and can be used in various fields of
organic analysis. (I have successfully used it to determine N-chloro-imids in the past.)
Bedlasky - 8-10-2022 at 14:25
Thanks, Pumukli!
Yeah, analytical chemistry isn't often discussed here. I find bromatometry very useful method. I tested it with many substances like Sb(III), Sn(II),
hydrazine, ascorbic acid, sorbic acid... Sorbic acid need long reaction time, for unsaturated carboxylic acids it is faster and more accurate use
simple NaOH titration in alcoholic environment, but it will do the job. Other mentioned substances can be quickly determined by direct titration, no
need for indicator (except Sb(III)). I plan to try titration of unsaturated ketones in chlorinated solvent by BrCl or IBr, I am really curious how
well it will go.
Pumukli - 24-11-2022 at 11:22
I tried to adapt this method to the determination of p-methoxy-phenol (PMP), but it seems to be not as simple and elegant as I hoped initially.
I encountered problems in the end, similar to the reported problems with N-alkyl-anilines. The reaction mixture (RM) fairly quickly regenerated iodine
and yellowed then turned to tea color again. Tomorrow I'll try to play with it more.
So far I noticed that after the bromination step the RM acquired a faint yellow colour. This yellowness is not exclusively the colour of excess
bromine though, because I could not remove it entirely with metabisulfite treatment. I think it is the inherent colour of the dibromo-methoxy-phenol.
And I think a dibromo compound is the result of the bromination because the end point of the weighted sample titration was close to the expected end
point of that stoichiometry. 
Now I'm speculating that this yellowness -if stable long enough- may open the way to VIS photometry as well. (Original PMP is colourless in solution
and I don't have a UV photometer to catch it without "derivatization", but have a VIS one and it might be good for the brominated product.)
On the other hand I might get away with an even simpler Fe3+-complex of this phenol (PMP) if I want to overcomplicate things with VIS photometry...

Direct bromatometric titrations
Bedlasky - 7-1-2024 at 19:06
Bromatometry can be very useful for quick determination of certain reducing compounds. Direct titrations can be employed with or without indicator.
Methyl orange and methyl red are usually used as indicators - these indicators are non-reversible (once they are oxidized, they can't be simply
reduced). Reversible indicators can be used if needed.
First excess of bromine formed in the solution can be used for end point determination if you titrate colorless substances.
Here I describe both methods of direct bromatometric titrations, namely:
Determination without an indicator (hydrazines, Sn2+, ascorbic acid)
Determination with methyl red/orange as indicator (Sb3+)
Chemicals used
0,1M KBrO3 (this needs to be exact concentration, mine was 0,1000 mol/l)
0,55M KBr
1+1 HCl
SnCl2.2H2O
Ascorbic acid
N2H5HSO4
Sb2S3
Determination of Sn2+/hydrazines
3Sn2+ + BrO3- + 6H+ --> 3Sn4+ + Br- + 3H2O
3N2H5+ + 2BrO3- --> 3N2 + 2Br- + 2H+ +
6H2O
Cca 2x0,68 g of SnCl2.2H2O or 2x0,2 g of N2H5HSO4 are quantitatively transfered in to two titration flasks and 2x20 ml of 1+1 HCl is added. Solutions
are titrated with 0,1 M KBrO3. You can observe yellow colour in the flask that quickly dissappear. This is colour of bromine that form when bromate
react with bromide formed during titration. As you progress the titration, yellow color starts to dissappear more slowly. Titrate slowly until the
yellow color remain persistent. This marks the end point of titration.
This method can be also used for determination of organic hydrazines (according to literature, I haven't test it yet).
Determination of ascorbic acid
5Br-2+ + BrO3- + 6H+ --> 3Br2 + 3H2O
C6H8O6 + Br2 --> C6H6O6 + 2HBr
Cca 2x0,53 g of ascorbic acid are quantitatively transfered in to two titration flasks and 2x20 ml of 1+1 HCl + 2x10 ml of 0,55M KBr are added. The
rest of the titration is the same as in the case of Sn2+/N2H5+.
In this case KBr must be added in advance, because ascorbic acid doesn't have well defined reaction with bromate (unlike Sn2+ or hydrazine). So
bromate must firstly react with bromide to form bromine, which react with ascorbic acid in 1:1 stoichiometry to form dehydroascorbic acid and bromide.
Reaction of ascorbic acid with bromine is fast, so ascorbic acid can be titrated directly (unlike other organic compounds).
Determination of Sb3+
3Sb3+ + BrO3- + 6H+ --> 3Sb5+ + Br- + 3H2O
Because colorless Sb3+ is oxidized to yellow Sb5+, you must use indicator for end point determination.
Commercial Sb2S3 already contain some Sb(V), because when I dissolved it in HCl, resulting solution already have yellow colour. So it was not the best
choice for testing this method, I should use KSb tartrate, but method work quite well.
1,2 g of Sb2S3 is dissolved in 25 ml of 36% HCl and boiled for a while to get rid of all H2S. Solution is filtered directly in to the 50 ml volumetric
flask, beaker and filter paper rinsed with 1+1 HCl. Then fill volumetric flask to the mark with distilled water, stopper it and shake properly. 2x15
ml from the stock solution are added in to two titration flasks and 2x20 ml of 1+1 HCl are added as well to dilute thing down little bit. Two drops of
methyl red or methyl orange indicator are added and titration flasks are heated to cca 60°C. Solutions are then titrated with 0,1M KBrO3. End point
is when solution became yellow (not orange) - this is colour of [SbCl5]2- ion. Titration proceed at 60°C because reaction of methyl red/methyl orange
with first excess of bromine is quite slow at room temperature and it is easy to overtitrate solution. Still, be patient and titrate slowly because
reaction of indicator with bromine still take little bit of time (few seconds).