From www.azom.com
____________________________________________
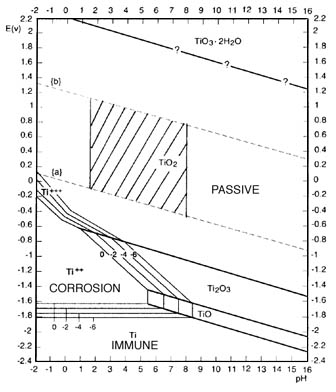
Oxide Film Growth
The oxide film formed on titanium at room temperature immediately after a clean surface is exposed to air is 12-16 Angstroms thick. After 70 days it
is about 50 Angstroms. It continues to grow slowly reaching a thickness of 80-90 Angstroms in 545 days and 250 Angstroms in four years.
The film growth is accelerated under strongly oxidizing conditions, such as heating in air, anodic polarization in an electrolyte or exposure to
oxidizing agents such as HNO3, CrO3 etc. The composition of this film varies from TiO2 at the surface to Ti2O3, to TiO at the metal interface.
Oxidizing conditions promote the formation of TiO2 so that in such environments the film is primarily TiO2. This film is transparent in its normal
thin configuration and not detectable by visual means.
A study of the corrosion resistance of titanium is basically a study of the properties of the oxide film. The oxide film on titanium is very stable
and is only attacked by a few substances, most notably, hydrofluoric acid. Titanium is capable of healing this film almost instantly in any
environment where a trace of moisture or oxygen is present because of its strong affinity for oxygen. Anhydrous conditions in the absence of a source
of oxygen should be avoided since the protective film may not be regenerated if damaged.
__________________________________________
Dann2 |