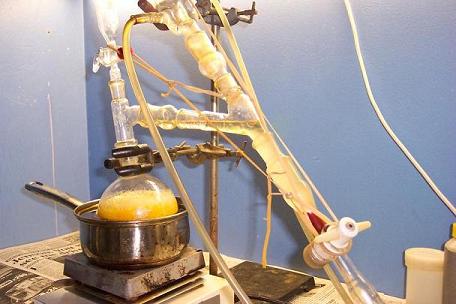
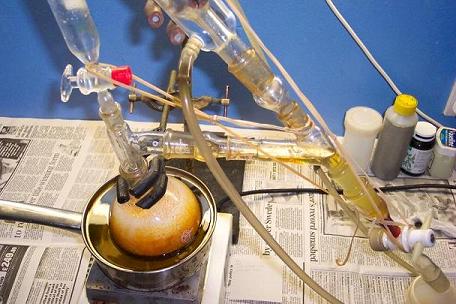
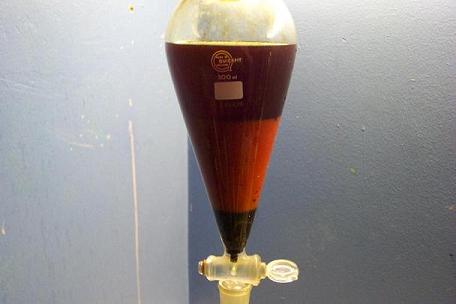
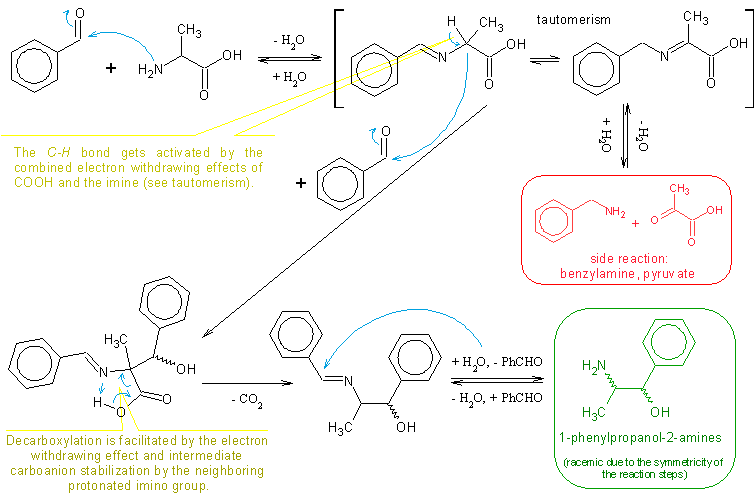
Originally posted by Maja
From one forum, one guy told that benzaldehyde can be replaced with other aldehyde as piperonal to get MDPPA and then to treat it same way as PPA
freebase to get P2P,but in this case we would get MDP2P. Maybe you have tried it and/or have refs / patents ? I WOULD be very grateful
|