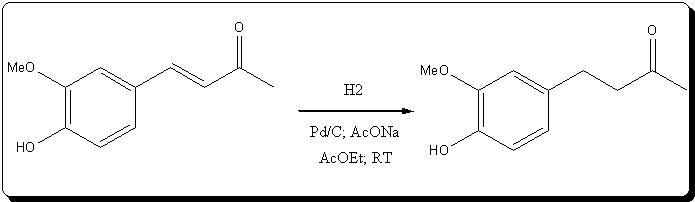
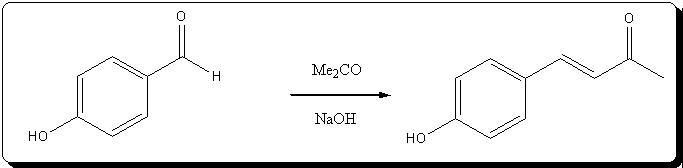
Anisaldehyde (4-Methoxybenzaldehyde [2])
A freshly prepared and stirred solution of 30 mL concentrated sulfuric acid in 150 mL water was allowed to cool down to 30° C, and anise oil (9.8 g)
was added. A total of 25 g sodium bichromate was then added, at such a rate that the reaction temperature remained between 35-40° C. The reaction
mixture was extracted four times with toluene (75 mL each), and the combined organic phases were washed twice with 5 % NaOH (100 mL each), and once
with water (100 mL). The organic phase was evaporated to about 20 mL, and anisaldehyde was then isolated as its bisulfite adduct. The yellow
precipitate was washed with an EtOH/ether (1:1) mixture until the precipitate's color turned white (that is, similar to the bisulfite adduct generated
from commercially available anisaldehyde). Setting the anisaldehyde free resulted in 4.9 g of a yellow oil with a pleasant odor. The mass spectrum was
in agreement with an authentic sample. Anisaldehyde was the main product (95 % by GC/MS), but several minor impurities (not further identified in this
report) were noted. |


 .
.