JnPS
Hazard to Self

Posts: 90
Registered: 29-7-2016
Location: PA, USA
Member Is Offline
Mood: Umpolung
|
|
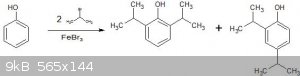
At home Aromatic Substitutions
I want to perform a Friedel-Craft's alkylation, but I'd like some advice from anyone who may have done one before. I'm trying to decide whether to use
a bromo-alkane &FeBr3 or a chloro-alkane &FeCl3. Are there any pros/cons to using one over the other?
Also, would anyone have a prep for FeBr3?
I found threads on FeCl3 but I couldn't find anything on FeBr3, if I missed it just give me the ol' UTFSE and I'll go back through and try to find it.
The file attached is the reaction I want to perform, obviously the bromine's would be replaced with chlorine's if I go that route. I'm assuming the
procedure is just put the reactants in a flask and reflux, but how would I separate the products from unreacted starting material? And any ideas on
separating the two isomers produced?
[Edited on 22-9-2016 by JnPS]
[Edited on 22-9-2016 by JnPS]
|
|
|
Texium
Administrator
Posts: 4515
Registered: 11-1-2014
Location: Salt Lake City
Member Is Offline
Mood: PhD candidate!
|
|
You could generate the FeBr3 in situ using iron and bromine. You won't need that much of it. Add a bit of iron to the solvent that you're
going to use in the reaction, and then drop in enough bromine to react with most but not all of it. Once the bromine color fades it should be ready to
go for the reaction.
|
|
|
Metacelsus
International Hazard
Posts: 2531
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
It probably would also work fine if you used iron bromide with a chloroalkane, or iron chloride with a bromoalkane. Isopropyl bromide will be more
reactive than isopropyl chloride (and also somewhat easier to deal with, due to its lower volatility.)
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
You don't need to use FeBr3 . You can do the alkylation using isopropyl alcohol and and an acid (ex- 85% H3PO4 )
http://www.mhhe.com/physsci/chemistry/carey/student/olc/ch24...
another method from isopropyl alcohol - http://pubs.acs.org/doi/abs/10.1021/jo01310a013
[Edited on 22-9-2016 by CuReUS]
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
Propofol is pretty nasty stuff, just so you know: that dose to response curve doesn't really instill confidence in me.
A major problem with Freidel Crafts in general is overalkylation. The alkyl groups donate electron density to the ring, and so after the first
substituent is added, the addition of a second or even a third group is favored. Thus, if you are looking for a disubstituted product, expect a lot of
trisubstituted material as a contaminant. In industry, they get around this by using a massive excess of benzene in the vapor phase, but this isn't
practical on a small scale.
I can't say much about the catalyst, since I've never done a Freidel Crafts before, but I don't think it will be too much more than treating some iron
wool in DCM with bromine solution in DCM or chlorine gas: the DCM should act as a diluent and a thermal control agent to keep things from going out of
hand. The alkyl halide is as trivial as heating sulfuric acid, a bromide salt, and isopropanol together and distilling-- isopropyl bromide has the
lowest boiling point and comes off first. Mixing halides shouldn't cause problems either, so the chloride works too.
Finally, if you are actually making propofol, what is stopping the isopropyl group from landing para to the hydroxyl? This should actually be favored
in some sense, since the para position has the least steric strain for the somewhat bulky isopropyl group. If you want a decent yield of product, you
will probably need to protect that position with something. I suspect sulfonation should work decently, but I'm not too sure.
|
|
|
clarichem
Harmless
Posts: 2
Registered: 17-9-2016
Member Is Offline
Mood: No Mood
|
|
As Cryolite mentioned, propofol is not the kind of stuff you would want to try using if you had nicked it from a hospital, trying to use your own
homemade stuff would be suicide, and Cryolite is right about over alkylations and needing a meta directing group on the para position, sulfonation
would work but is difficult to remove, I would go with a nitro group. Also Friedal-Crafts alkylations tend to only work with tertiary alkyl halides -
not impossible, but expect a crap yield.
Making propofol from phenol with a high yield and clean product will require multiple steps, some requiring difficult to obtain/prepare reagents like
methyl grignard, ethanoyl chloride and hydrogenation catalysts and will demand air-sensitive techniques - not the kind of thing you want to do unless
you really know what you are doing and are in a proper lab.
That said, If you are hell bent on doing it I can sympathise (I loved doing experiments at home before going to University) so I can advise on a
better synthesis if you are not swayed by the whole 'if you do it you are probably breaking the law and if you use it you are likely going to
euthanise, not anesthetize yourself/get high' thing.
|
|
|
JnPS
Hazard to Self
Posts: 90
Registered: 29-7-2016
Location: PA, USA
Member Is Offline
Mood: Umpolung
|
|
Some disclaimers: Yes I want to make propofol, no I will not be using said propofol, I just want to explore the chemistry. I'm going to try and make
it, take an IR, and then dispose of it. Also I checked and propofol isn't a a ClassI-IV controlled substance and I doubt its a class V, I know
fospropofol would be illegal so obviously I will not be putting a phosphate on the thing. I keep a word document with every ClassI-IV substance to do
a quick ctrl+f check every time I want to make something new. Class V substances usually are notorious enough that while I'm researching it the
illegality is usually explicitly stated before I pursue it further.
|
|
|
clarichem
Harmless
Posts: 2
Registered: 17-9-2016
Member Is Offline
Mood: No Mood
|
|
Alright, fair enough.
Your best starting material is paracetamol, it has the OH and a meta-directing group para to the OH - perfect!
You will then want to acylate that with 2 equivalents of acetyl chloride with anhydrous AlCl3 as a catalyst (use 1 mol.eq.) and DCM as a solvent. An
hour on reflux should do the trick, given that paracetamol is dirt cheap you can always save the mixture and add more paracetamol.
After purifying your product, you will then want to hydrolyse that amide, dilute HCl or NaOH will work fine, reflux in a 2 mol solution for an hour,
the product should be more soluble so stop when it's clear (assuming the reactant was not very soluble)
Now for a more challenging reaction, conversion of the amine to a diazonium salt. These are very temperature sensitive and decompose to form an OH
above 5 celsius, awful, so all of this needs to be performed in an ice bath.
Start by dissolving your product in a strong HCl solution, 6M should be good then add 1.1 mol.eq. NaNO2, sodium nitrITE, NOT nitrATE! (this can be
made by thermally decomposing NaNO3) and stir for 1-2h, keeping the ice bowl topped up. Now add 1 mol.eq. H3PO2 - unfortunately if you are an American
this stuff is DEA List 1, wiki has a preparation. I will post if i can find an alternative but it's unlikely there will be anything accessible. If you
do have it, once you add it you will have replaced the diazonium with a hydrogen.
Now for a grignard reaction. This reaction is moisture and air sensitive, not to mention is used in illicit drug manufacture... if you are caught,
saying "i wasn't making meth! i was making propofol!" is unlikely to help your case, and i say caught because get this wrong and there will be a fire,
do it on a small scale. You will need a oven dried product, as well as anhydrous diethylether, oven dried quickfit glassware and need to make methyl
iodide - this stuff is seriously toxic. It is made by reacting methanol with hydroiodic acid (another DEA list 1 reagent) which is made by reacting
NaI/KI with conc phosphoric acid and distilling. Once you have made the methyl iodide, dry it and put it in your dry flask, add dry ether and dry
magnesium shavings (freshly washed with acid to expose the metal surface) and add an iodine crystal. you should have a condenser with a drying tube on
the top of the flask and control the reaction with a ice bath. once it has died down you have prepared your methyl magnesium iodide in ether.
depending on the concentration, exposure to air will either just degrade it or ignite it! now add your product dissolved in ether SLOWLY, once added
reflux for an hour and then pour into dilute HCl, keep the top layer (ether) and evaporate to get your product
Assuming you have survived, dehydrate your product by heating it with conc sulphuric for a hour or so, then dilute, extract using DCM and evaporate.
Now for an even harder reaction, hydrogenation. You need to get hold of Palladium on carbon for this, or make it. Nickel only works are high pressure
and temperature. add your dry product to a dry flask and dissolve in methanol/DCM mixture, add the Pd/C catalyst (10mol% of 10%Pd/C is standard) and
then apply a vacuum to the flask for a few seconds then introduce hydrogen gas from a balloon - if you dont get the air out and add hydrogen, boom.
stir for a hour then filter and evaporate the solution to get your propofol
Obviously this is a mammoth undertaking for someone without access to a proper lab, lots of specialised equipment will need to be designed and
improvised and many dangerous/restricted reagents will need to be prepared.
There are probably easier synthetic strategies, but it is 11pm here in the UK and I designed this synthesis quickly as a rough guide to show you what
you are up against.
If you are still prepared to do this, knowing that if your practical skills are not great, you will get little to no yield by the end and perhaps even
take a trip to hospital then I am willing to help you further - I can do literature searches and optimise the reactions to what reagents you have/can
make. I am happy to do this simply as an academic exercise (my degree specializes in organic synthesis and drug design) and if you want to attempt,
do so at your own risk.
|
|
|
Maroboduus
Hazard to Others
Posts: 257
Registered: 14-9-2016
Location: 26 Ancho Street
Member Is Offline
Mood: vacant
|
|
Sodium nitrite is no problem in the USA, It can be bought by the pound on Amazon. We put it in the food we eat on St Patrick's day. But we don't have
a Grinard reagent holiday, unfortunately.
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
Aren't acylamino groups ortho para directing? That synthesis won't work.
|
|
|
Maroboduus
Hazard to Others
Posts: 257
Registered: 14-9-2016
Location: 26 Ancho Street
Member Is Offline
Mood: vacant
|
|
Nice catch, Cryolite.
By the way, are you from Ivittuut?
BTW, I suspect that there's a sneaky way you could get there from potassium salicylate. But the penultimate step might not work and I have some
scruples about going into the details since I don't want to be responsible if somebody else sees this and kills himself by making and using the
product. This stuff might not be regulated, but it sounds like bad news.
EDITED to correct my crappy spelling. At least what I could catch with my crappy proofreading.
[Edited on 23-9-2016 by Maroboduus]
[Edited on 23-9-2016 by Maroboduus]
|
|
|
JnPS
Hazard to Self
Posts: 90
Registered: 29-7-2016
Location: PA, USA
Member Is Offline
Mood: Umpolung
|
|
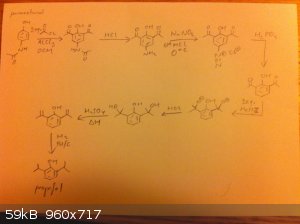
Would this not work? It looks good on paper, but is there a practical problem in doing it this way?

|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
How do you ensure you nitrate the para position and not the ortho one? Further, what is stopping dinitration? I realize nitro is far more electron
withdrawing than bromine, but phenol almost immediately decolorizes bromine water due to rapid trisubstitution.
The reason I suggested sulfonation is because unlike most forms of electrophilic aromatic substitution, sulfonation is reversible with heating. If you
try and sulfonate phenol at room temperature you get mostly ortho hydroxy benzenesulfonic acid( the kinetic product), but with heating on a water bath
you get mostly para hydroxy benzenesulfonic acid ( the thermodynamic product, due to sterics). This can then be alkylated and desulfonated without too
much trouble, at least on paper. (You may need to worry about the formation of the sulfone, though. The reaction conditions are remarkably similar to
the ones used to make bisphenol S, after all. An excess of sulfuric acid should help mitigate this.)
|
|
|
JnPS
Hazard to Self
Posts: 90
Registered: 29-7-2016
Location: PA, USA
Member Is Offline
Mood: Umpolung
|
|
So sulfonate, alkylate, desulfonate...sounds simple enough, won't have to worry about trying to get any HPA. I'll start prepping and get back to SM
with the results in a week or two, thanks guys.
As a side note I saw on chemspider that 2,6 diisopropyl phenol is a solid where as 2,4 diisopropyl phenol is a liquid at room temp, what's the theory
behind the dramatic difference in properties between the two?
Well I assumed this based on chemspider supplying a melting and boiling point for propofol but only a boiling for the 2,4 isomer
[Edited on 23-9-2016 by JnPS]
|
|
|
clearly_not_atara
International Hazard
Posts: 2694
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Acetanilides can be converted to N-nitrosoacetanilides with N2O3 and cleaved to the unsubstituted arene. So deprotecting paracetamol is easier than it
sounds; no hydrolysis required. See:
http://pubs.acs.org/doi/abs/10.1021/ja01069a062
The FC-alkylation of paracetamol with isopropyl bromide and FeBr3 should proceed rapidly. Deprotection with N2O3 followed by heating in diethyl ether
gives the target compound.
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
atara, I still don't think acylamino groups give you the selectivity you need. Although hydroxyls are stronger directing groups than acylaminos, the
effect should be significant enough to produce a lot of byproducts.
On the other hand, another route which might be of some use would be to start from salicylic acid, rearrange to p-hydroxy benzoic acid by heating,
esterifying with isopropanol to isopropyl paraben, alkylating with isopropyl bromide to isopropyl (3,5-diisopropyl-4-hydroxy benzoate), and then
hydrolyzing the ester and decarboxylating to propofol.
|
|
|
Dr.Bob
International Hazard
Posts: 2659
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: No Mood
|
|
A simple search yields many routes with yields, procedures, and more.
https://www.google.com/#q=propofol+chemical+synthesis
There is a simple one designed for lab scale in
http://www.umich.edu/~chemh215/CHEM216/HonorsCup/HC%20250-IV...
but it appears to be a plan rather than a detailed prep.
Then there is a real manufacturing prep at
http://pubs.acs.org/doi/abs/10.1021/op400300t
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
Nice to see that my paraben based route was somewhat practical 
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
it won't be an acylamino group anymore after you react it with N2O3
[Edited on 24-9-2016 by CuReUS]
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
Oops, guess I misread. Regardless, nitroso groups are electron withdrawing, but they are ortho para directing like the halogens.
[Edited on 24-9-2016 by Cryolite.]
|
|
|










