Klaus
Harmless

Posts: 10
Registered: 27-7-2018
Member Is Offline
|
|
P2NP synthesis/crystalization problem
1. 150ml of saturated NaHCO3 water solution was prepared.
2. 36ml of lab grade benzaldehyde was poured in the solution with a syrenge and vigorously stirred.
The beaker was covered with another beaker to limit the oxygen exposure and was left for 2-3 min
for layers to separate. The upper layer was decanted and was about 29ml.
Another 4ml or so were recoverable. - that should be approx 3ml of benzoic acid. The pH of the NaHCO3
solution appear to be the same when measured with cheap pH paper.
3. To a 500ml RBF was added 80ml GAA + 15g very wet ammonium acetate (can be pressed
against a piece of toilet paper to remove at least some of the excess water).
The mixture was shaken for about 5-10 minutes until the acetate dissolved.
20ml of the washed benzaldehyde ware added to 22ml room temp nitroethane.
This was mixed and added to the GAA solution. The solution was mixed and
put on reflux on water bath, water temp lower than boiling, probably around 80 C.
GAA will condense at low temp so a condenser is hardly neccessairy, but is used anyway.
What I mean is, most of the liquid condenses on the vessel walls, so probably a clandestine
setup of some kind with some ice will do just fine.
It is a good idea to put a ball of cotton wool or toilet paper in the top of the condensor,
so water from air won't condense and drip down in the reaction vessil, but is loose enough
that pressure won't build.
Stoped after 3 hours. Color is light orange
250ml ice-cold d.water was poured on top. 3 layers formed - bottom darker orange about 25 ml
and opaque white aqueous on top. on top of the white layers is a spot of bright yellow
organic stuff, a bit of which is also floating in the white layer in the form of small yellow
bubbles.
4. The bottom organic layer was separated and put in fridge for 2 hours.
The ecess watery/acid stuff froze and was removed but not much else happened.
5. Out of the frige, the dark-orange liquid was washed with 100 ml of sat Na2S2O5 water solution.
Not much happened at first, probably because of the lower pH(about 6), but after 5 min some cristals started forming.
With stirring, much more crystals formed, and after about 10 mins we had some thick formable goup
full of crystals with the consistency of orange juice pulp.
It was scooped and was about 35ml. This was washed 2 times with water until benzaldehyde adduct
crystals dissolved. The remaining orange liquid was collected(about 15ml), ice was added, but yet no
clue of P2NP crushing out.
6. This was washed with sat Na2S2O5 water solution again and nothing happened, so the orange liquid was
transferred to a test-tube, but then some adduct crystals was observed in the test tube have started forming,
so some more sat Na2S2O5 water solution was added to the test tube and it was caped and shaked vigorously
and now looks just like a thick orange juice full of smallest bubbles that don't disappear at all which I
like to shake or just look at, and think to my self "look what you have done". It is quite prety actually.
But seriously, I attempt this reaction for 6 or 7th time following different protocol each time
and fail misserably to produce P2NP crystals every time. Please help.
And how about the orange stuff, do you think it still contains some recoverable P2NP?
P.S. Please, excuse my typos and stuff, but most of this was recorded during the reaction.
Also, please excuse my orange-juice metaphors
|
|
|
zed
International Hazard
Posts: 2277
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Try a better procedure.
https://www.thevespiary.org/rhodium/Rhodium/Vespiary/talk/fi...
Do you have a Soxhlet extractor or a Dean- Stark trap?
|
|
|
Klaus
Harmless
Posts: 10
Registered: 27-7-2018
Member Is Offline
|
|
Nope. I also don't have a microwave  I have a bottle of Di-N-Butylamine though,
but I'm using Ammonium acetate in GAA as it don't require a Dean- Stark trap. I have a bottle of Di-N-Butylamine though,
but I'm using Ammonium acetate in GAA as it don't require a Dean- Stark trap.
People report the Ammonium acetate to be really easy and forgiving, though a bit lower-yielding and producing a bit dirty product.
I suppose it is because of the water - my Ammonium acetate is not anhydrous. What product forms in the knoevenagel condensation in the presence of
water? Is it that the OH can't be dehydrated or what. My thinking was that I have some P2NP dissolved in unreacted benzaldehyde that I will eliminate
via the adduct(and then reuse), but it is probably not the case. I don't know...
I'm a bit tired right now, but I will explain in detail what I was thinking tommorow.
Why Soxhlet extractor btw?
[Edited on 28-7-2018 by Klaus]
[Edited on 28-7-2018 by Klaus]
|
|
|
zed
International Hazard
Posts: 2277
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Either the Soxhlet extractor or the Dean-Stark trap, may be used to drive the reaction, in the aforementioned procedure, by removing water as formed.
Use the search engine. Lots of different procedures should be recorded there, that work well to produce these materials.
Some members here, have used phenethylamine or other PRIMARY amines as a catalysts.
https://www.sciencemadness.org/whisper/viewthread.php?tid=76...
[Edited on 28-7-2018 by zed]
|
|
|
Corrosive Joeseph
National Hazard
Posts: 915
Registered: 17-5-2015
Location: The Other Place
Member Is Offline
Mood: Cyclic
|
|
This is NOT the right forum for this but before this goes to detritus...........
Di-N-Butylamine won't work and dean stark trap is not needed.
Ammonium acetate isn't great either..........
Best catalysts are acetate salts of n-Butylamine and methylamine.
Taken from another forum.........
"They use Toluene as the solvent when using a dean stark trap. The Azeotrope would boil over at 85C and seperate in the trap.
Honestly, it is the most outdated and backwards low yielding way to perform this reaction. Do it in Ethanol and use GAA to neutralize your catalyst
and things will go very well."
"If you heat it for two-four hours at 100°C in ethanol, you will certainly have a good product, just don´t let the color get too a deep yellow(even
that is too dark!).
But if you let it get red, you will receive some tar as product, making it hard to crystallise.
If you use ethanol it will give the best results and easiest crystallisation, followed by IPA, MeOH...... and toluene is the worst of them all.
Even if standing days or weeks at -18°C, it won´t crystallise out, had to remove half the solvent and wait a few days until a giant crop of crystals
fell out.
The reaction will complete when standing at room temperature.... at least three weeks though. Just put in a dark, warm place....... "
Crystallization -
"Minimum of a 3.5 to 1 ratio is what might end up being used, as in 3.5L Ethanol to 1kg P2NP, or 350-400mL Ethanol to 100g P2NP. There's no set
formula. The re crystallization is notoriously easy as EtOH is the perfect solvent for this particular compound. Using more EtOH is not a problem and
can make things easier.
DON'T using boiling EtOH! Always think about the melting point of the solid you want to clean up. In this case, you will ruin it by using boiling
solvent. Start at 40C-45C under heat and mag stirring, add your P2NP in small increments until it dissolves. Keep adding it slowly until everything is
added. Bring it up to heat until everything dissolves. Use a squirt bottle to spray the sides of the beaker as crystals form. Usually around 50C is
enough Heat for everything to dissolve. If not, add more Ethanol not more heat!
Set it aside at room T and cover undisturbed. Filter once crystallization is full avoiding mixing the crystals with the mother liquor. Wash with fresh
EtOH during vacuum filtration. Place the mother liquor and washes into freezer to see if any more crystals form. Be aware the second crop of crystals
might not be of same quality and may need an additional re crystallization."
/CJ
[EDIT] - I will never post on this reaction again. Period
[Edited on 28-7-2018 by Corrosive Joeseph]
[Edited on 28-7-2018 by Corrosive Joeseph]
[Edited on 28-7-2018 by Corrosive Joeseph]
Being well adjusted to a sick society is no measure of one's mental health
|
|
|
Chemi Pharma
Hazard to Others
Posts: 349
Registered: 5-5-2016
Location: Latin America
Member Is Offline
Mood: Quarantined
|
|
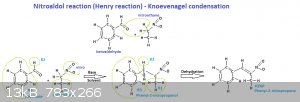
I think your problem is the work up you are doing.
My comments: don't use NaHCO3 to try purify your benzaldehyde from benzoic acid. Any residual benzoic acid that exists in your benzaldehyde don't hurt
the reaction at all but, otherwise, carbonate or bicarbonate impurities favours the formation of phenyl 2-nitro 1 propanol instead phenyl
2-nitropropene like this:

Although PIKAL (Alexander Shulgin) used ammonium acetate as a cataliser in the synthesis of 2,5 DMA, like below, you'd better use some kind of amine,
like cyclohexylamine or n-butilamine that usually give higher yields than ammonium acetate, like in the synthesis of MDA, also below:


Rhodium brings three different ways to do this Henry Reaction with n-butilamine, cyclohexylamine or ammonium acetate as a cataliser, as seen below:

Also, I recovered a video made by TDC user, deleted from his You Tube Channel and uploaded by me at my Bit Chute Channel, on how to synthesise P2NP
with n-butilamine as a cataliser, ethyl alcohol as a solvent, benzaldehyde and nitroethane. No acetic acid, no ammonium acetate, no NaHCO3 and no
Thiosulfate at all, like you did. By the way I still don't understanding why you have used Thiosulfate in your work up. No sense for me. In the video
TDC recristalized the P2NP from hot methanol.
Here's the link for the video: https://www.bitchute.com/video/W0pd0RzI51a3/
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|

|
|
|
Klaus
Harmless
Posts: 10
Registered: 27-7-2018
Member Is Offline
|
|
I know that all primary and most secondary amines will work. Why di-n-butyl won't? All that is needed is a weak base, and it's pKb is similar to that
of the n-butylamine. And btw, how do you tell which amine will do and which won't? And how do you calculate the exact ammount needed, as it varies for
each catalyst?
Why I would want to buy expensive, poisonous and suspicious amine when I can use dirt cheap innocent Ammonium acetate, and if a bit lower yielding -
so what?
I used the Thiosulfate because I thought that I have P2NP dissolved in Benzaldehyde that won't crystalize ever.
The Thiosulfate reacts with the excess benzaldehyde to form an adduct that is water-soluble and is just washed with pure water then.
[Edited on 28-7-2018 by Klaus]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|

|
|
|
Klaus
Harmless
Posts: 10
Registered: 27-7-2018
Member Is Offline
|
|
[Edited on 28-7-2018 by Klaus]
|
|
|
j_sum1
Administrator
Posts: 6219
Registered: 4-10-2014
Location: Unmoved
Member Is Offline
Mood: Organised
|
|
[moderating]
JJay, I have the same facial expression as your cat. P2P has no legitimate uses and the mere presence of this thread makes me uncomfortable.
Having said that, the OP does fall on the correct side of board policy.
Quoting Polverone in a mod discussion on board policy wrt drug threads
| Quote: | | You can discuss the chemistry of all of them, as long as you communicate like a scientist and make it clear you've done your homework.
|
and
| Quote: |
no SWIM or other cook-slang allowed, no questions that point to an obvious commercial interest (e.g. "how do I scale this synthesis up to 1 kg?"), and
people asking questions must have shown that they put some of their own effort into answering the question first. |
Since the question related to a crystallisation problem I let it remain.
Since this appears to be adequately answered by zed (use Soxhlet or Dean Stark), by Corrosive Joseph (solvent choice and crystallisation) and by Chemi
Pharma (work-up procedure) I think it is probably time to close it down.
If the OP finds that the references and advice provided by those three posters is inadequate then he is welcome to ask further questions with the
reminder that
(a) This is a chemistry forum of a general nature with no specific interest in drug synthesis and most members having a general disdain for kookery.
(b) Questions that focus on chemistry are welcome. Questions that amount to spoon-feeding for drug recipes; where the only practical application is
the clandestine manufacture of illicit substances: these questions are not welcome.
[/moderating]
|
|
|
j_sum1
|
Thread Closed
28-7-2018 at 00:20 |









