| Pages:
1
2 |
len1
National Hazard

Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Benzotrichloride, Benzoyl Chloride, and Phthalyl Chloride - Illustrated Practical Guide
This article is part of a series of Illustrated Key Syntheses in Chemistry which I intend to post, with the purposes and guidelines outlined in the
thread of the same name in the General Chemistry section. I shall attampt to ensure that all the relevant information is contained in a single post.
If sufficient reason arises for something to be changed or edited, I shall edit the entire post.
Aim
As has been mentioned in the article on the synthesis of thionyl chloride, simple methods for manufacturing alkyl and acyl chlorides open the door to
the synthesis of a large range of organic compounds. Although thionyl chloride is an excellent reagent for this purpose its main disadvantage so far
has been the laborious nature of its preparation, in particular the need to handle large amounts of SO3 and the potential danger this poses should
accidents occur. However, it is well known that acid chlorides can be converted to one another by acting on with the corresponding acid, the
equilibrium being progressed if the more volatile chloride is continuously removed.
The road to acid chlorides for the amateur chemist is provided by the well known property of benzotrichloride to generate acid chlorides by its action
on water, organic acids, or acid anhydrides (for the earliest work see Deutsch Patent 11494, 1879). It can also generate acid anhydrides by acting on
organic acid salts, this however is not the preferred way for the synthesis of anything but benzoic acid anhydride due to the formation of mixed
anhydrides as a byproduct.
I have already demonstrated that benzotrichloride itself can be easily prepared in good yield by blue-light mediated chlorination of toluene in the
article on benzaldehyde synthesis. In that work, benzotrichloride was evidenced only by its ~10% contribution at 629 cm-1 in the IR spectrum of the
isolated benzal chloride, and was thus an undesired impurity. The ability to continue the chlorination to actually achieve a good yield of this
compound has up to this point been subject to severe doubt, due to the existance of quite contradictory statements on this point in the literature.
Two antigonistic viewpoints to the success of this synthesis raised the following doubts
Free-radical chlorination becomes incredibly inefficient in the last stages of chlorination, requiring such exceedingly large amounts of excess
chlorine as to make the process unviable in a batch amateur setup.
By-products in the form of ring-chlorinated compounds and intractable polymerisation tars form in greater and greater amounts as chlorination is
continued. The latter can arise due to the continuous increase of the b.p. of the chlorinated mixture and the increase in polymerization tendency of
chlorinated toluenes with temperature, the former due to the decrease in efficiency of free radical chlorination as a consequence of the reaction rate
constant for the third chlorination being a factor of ~25 times smaller than for the first chlorination, while the reactant concentration also
continuosly drops. These factors can potentially make the yield unperspective.
Other sources, such as the Kirk-Othmer Encyclopedia of Chemical Technology mention that chlorination is carried out to almost 100% benzotrichloride in
industry, indicating that such an outcome is at-least possible, although no details of the conditions under which such conversions obtain are
mentioned.
Here I have definitively found that the chlorination of toluene is essentially complete, high-yielding, and free of any IR detectable impurity. The
excess of chlorine required for the entire procedure is 100% (this is based on TCCA used and not on chlorine generated, so this excess can be
decreased at the expense of longer process time) and these results can be obtained using simple equipment in a batch process. The chlorination time
from the benzal chloride to benzotrichloride, is not excessive, being only about 1/3 of the time needed to reach the benzal chloride maximum. If the
aim is benzoyl chloride, this clearly puts this method ahead of the chlorination of benzaldehyde route, which according to the literature requires a
dozen or so hours.
Moreover I have found the conversion of the trichloride to acid chlorides, and the interconversion of acid chlorides to be very pure and high-yielding
processes. These processes, outlined in 30's literature (H.Brown JACS Vol.60 p. 1326, and L.P.Kyrides JACS Vol. 59 p. 206), have given me equivalent
yields as for the original authors and in some instances cleaner products.
Findings
86% yiled of benzotrichloride based on toluene in a two-step procedure
Benzotrichloride contains no foreign IR peaks (purity > 95%)
First chlorination of 2.2mol of toluene was carried out at b.p. in 1L flask, lasted 7hrs with 1kW high-pressure Hg illumination at 8cm with fan
cooling, used 700grams TCCA and 875ml 30% HCl, produced 30gms (~15%) loss of product to polymerised tars, and 164gms weight gain, corresponding to a
composition of Ph-C-Cl2.54-H0.44 (with account for polymerisation loss), which was supported by Beers law analysis of IR spectra giving a benzal
chloride/benzotrichloride ratio of 1:1 based on peaks at 586 and 629 cm-1. The chlorine excess based on this composition and the amount of TCCA used
is therefore 90% (190% of the required amount).
Second chlorination of 273gms of the distilled product from the first step carried out at a gradualy decreasing temperature range from 180C to
150C, took 3hrs, 150gms of TCCA and gave a weight gain of 37gms and a pure product based on IR. This gives, and is supported by a comparison of first
step data with overall data, a yield of effectively 100% for this step (within experimental error). The chlorine excess for this stage based on TCCA
is therefore 150%.
The chlorine excess for both steps combined in a 10 hour processes based on TCCA used is thus 100%. Note that this does not mean all this extra
chlorine has been generated - the TCCA was replaced as soon as a 20% or so decrease in chlorine evolution was evident. This is based on the cheapness
of the chlorine generating precursors. Very little chlorine was evident in the exhaust gases - except in the second step.
Benzotrichloride reacts rapidly at 100C, and effectively quantitatively with phthalic acid in the presence of zinc chloride to yield benzoyl
chloride and phthalic anhydride. 145gms (0.74mole) of benzotrichloride reacted in 1 hr with 112gms of 90% phthalic acid (0.61mole) - 10% phthalic
anhydride (0.08mole) in the presence of 10gms ZnCl2.(0.17 H2O) to yield 99.9gms (0.71mole) of benzoyl chloride a 96% yield - 4% lost most likely in
distillation - and phthalic anhydride. Weight loss of reactants was 50gms, which assuming was all HCl amounts to 1.37mole, suggesting a few grams of
benzoyl chloride was lost with the HCl.
All the benzoyl chloride distilled in a narrow range of 78-80C at pump pressure and was IR pure. The phthaloyl chloride distilled at 138-142C
at pump pressure as soft white crystals, and was almost IR pure. The pump pressure was indicated by a vacuum manifold gauge to be about (atmosphere -
27inch Hg) which evaluates to about 74mm Hg - however tabulated vapour pressures for both isolated compounds suggest the manifold gauge is inaccurate
by about 2inch Hg, and the actual vacuum was about 12mm Hg. (b.p. benzoyl chloride 78C at 12mm Hg, b.p. phthalic anhydride 140C at 12mm Hg). The
intermediate fraction was negligible (< 1gm).
Benzotrichloride in the amount of 84.1gms (0.43mole) was reacted with 60.3gms (0.41mole) of phthalic anhydride in the presence of 3.2gms
ZnCl2.(0.17 H2O) (0.004mole H2O) for 12 hours at 120-130C, as per Kyrides but with the reaction time cut in half and temperature raised by 10C. On
completion of reaction 54.1gms (0.385mole) of IR pure benzoyl chloride was recovered by fractional distillation through a Hempel collumn at reduced
pressure in the 80-82C temperature range. This amounts to a yield of 90%. 71.9gms (0.354mole) of IR pure phthaloyl chloride came over in the
132C-136C temperature range, with almost all the chloride coming over at 136C. This amounts to a yield of 86%. 3.4gms of substance, including some
HCl, was lost as vapour in the course of the reaction, 6.4gms was collected this time as a middle fraction in the temperature range 86C-132C, while
the remainder of yield remained asvpolymerization product in the flask, and some undistilled liquid in the Hempel collumn.
Any moisture in the ZnCl2 - which can not be removed by evaporation due to the formation of zinc oxychloride, zinc chloride being a moderately
strong Lewis acid - is removed at about 70 - 80C by the benzotrichloride with evolution of HCl and formation of benzoyl chloride. In the above case
the approximately 0.25gms H2O in the zinc chloride contributed to a 'loss' of about 2.7gms benzotrichloride in this way. The subsequently anhydrous
ZnCl2 in addition to catalysing the desired reaction forms sites for polymerization of the benzotrichloride as is evidenced by a brown-black surface
coating of the crystals, which increases with temperature.
Benzotrichloride was found to be a heavy slighly viscous liquid with a density of 1.36 (84.4gms in 62ml). It is initially yellow due to
dissolved chlorine, however on distillation it is almost colourless with a slight hint of yellow. The latter reappears on heating probably due to
formation of polymerization products and evolution of chlorine. It has a heavy sweetish smell somewhat reminiscent of chloroform, which gives one a
headache on any more than a brief exposure. The liquid fumes when a bottle is opened on humid days, however on days with humidity < 50% this was
not evident. Benzoyl chloride is a transparent liquid with a not-unpleasant pungent fruity smell which is also reminiscent of benzaldehyde, it is
however almost immediately lachrimatory, much more so than the chlorotoluenes. This is probably due to its greater polarity and so water solubility.
Phthaloyl chloride is a heavy transparent liquid with little odour, in ordinary vapour concentrations I did not have lachrymatory problems with it.
It is known, see Kyrides, that some acid chlorides can be used to synthesise thionyl chloride - which is an inorganic mixed anhydride. Since
benzotrichloride has been seen to act in a certain sense as an acid chloride (it converts acids to acid chlorides) it was reacted with a rapid stream
(~5 bubbles/sec) of dry SO2 for a 5 hour period at temperatures up to 165C. No traces of SOCl2 - only BzCl - was found in the exhaust gases. Raising
the temperature further, as per 200C employed by Kyrides, is counterindicated by the proximity of the b.p. and decomposition of the benzotrichloride,
as well as of the SOCl2. The later, rather than losses with SO2, I suggest contributed to the generally low yield of SOCl2 for this type of reaction
obtained by Kyrides. It is possible that SO2 is unreactive with benzotrichloride due to the low polarizability of the latter.
Theory
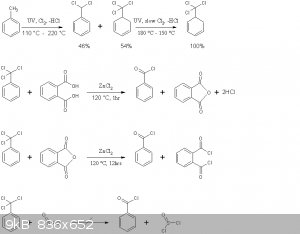
The reaction schemes of this practical are presented below
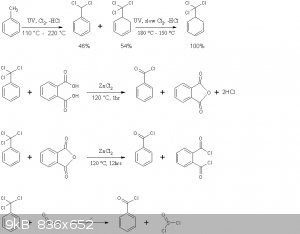
Note that benzoyl chloride rather than phthaloyl chloride is distilled off in the second reaction above. This is in accord with the general
principle seen in Brown's paper, where the equilibrium is shifted to the more volatile chloride by distillation. Moreover no mixed acid-acid choride
compound of the type Cl(CO)-C6H4-COOH is formed, the later immediately decomposing to HCl and phthalic anhydride.
As outlined in Deutsch Patent 11494 by Emil Jacobsen, benzyltrichloride in the presence of a weak Lewis acid, such as ZnCl2 hydrolyzes with the
stoichiometric amount of H2O according to
PhCCl3 + H2O -> Ph-(CO)-Cl + 2HCl
excess H2O leads to complete hydrolysis as in the benzaldehyde thread
Ph-(CO)-Cl + H2O -> Ph-COOH + HCl.
Moreover since the equilibrium of the latter reaction is essentially completely to the right, provided one does not err in the amount of H2O added
during benzoyl chloride synthesis by more than a factor of 2, no H2O will be present in the final product.
Interconversion of acid chlorides and anhydride formation
The benzoyl chloride formed in the first reaction can be reacted with many organic acids, as per the paper by Brown, provided its chloride is more
volatile, it can be distilled off. Thus
BzCl + AcOH -> BzOH + AcCl
However this is quite a disadvantageous use of benzotrichloride, and twice the yield of the acyl chloride can be obtained by by-passing Browns
procedure and using the direct action of benzotrichloride, as per Emil Jacobsens patent
PhCCl3 + AcOH -> BzCl + HCl + AcCl
BzCl + AcOH -> BzOH + AcCl
overall
PhCCl3 + 2AcOH -> BzOH + 2HCl + 2AcCl
For anhydride synthesis however, all anhydrides except those of benzoic acid and anhydrides of dibasic acids such as phthalic anhydride,
benzotrichloride should not act directly on the acid salt due to the following competing processes
AcONa + AcCl -> Ac-O-Ac + NaCl
BzONa + AcCl -> Bz-O-Ac + NaCl
BzONa + BzCl -> Bz-O-Bz + NaCl
all but the first yielding a byproduct. It is best to isolate the acyl chloride first using its volatility difference, and then act it on the
anhydrous acid salt.
Method
All operations described below were carried out in a fume hood, wearing a gas mask, and latex
gloves.
Ambient temperature in the lab 20C
Formation of benzal chloride/benzotrichloride 50% mixture - step 1
350 ml of commercial toluene was placed in a 1L flask and distilled - with the condenser jacket initially dry - with 202 grams (227ml) of an
intermediate fraction being collected, after 50ml or so of toluene has passed. The first fraction containing water, and the third fraction left in
the still containing xylenes and metal salts are combined and used subsequently to wash polymerization products from the reaction flask (they show
little solubility in other solvents).

Chlorinating apparatus with high-pressure Hg illumination is now assembled as per the benzaldehyde thread (joint grease must not be used as the
chlorotoluenes will attack it). Care is taken to locate the Hg lamp a minimum 8cm from the reaction flask (to minimise hot-spots) and slighly above
it - as after the solution has darkened due to formation of condensation products, much of the chlorination will occur in the vapour phase. This has
the double benefit of minimising ring chlorination. A slow air current from a desk fan is used to cool the lamp. The chlorine generator with its
adjustment valve is located as far as practicable from the reaction flask, with an indespersed shield to minimise radiation exposure during
adjustment. The entire fume-hood window is shielded with a heavy curtain screen during the reaction (beware that many materials will discolour). 5
moles of NaOH are placed in the scrubber vessel - sufficient to bind all the HCl generated, while forming an acidic NaCl solution at the end to
indicate the reaction rate at that point.

The 1L flask is heated at full rate on a 400W mantle and the course of the reaction is followed by the rising temperature of the reflux. An HCl drip
rate of 1 drop/2 sec on 200gms of pulverised TCCA is used, and as soon as the rate of temperature rise shows a decline (normally after 250 ml 15% HCl
had been added) the TCCA is changed. The initial temperature rise is rapid compared to that later on, due to the progressing diminution of the
temperature difference between the b.p.'s of chlorinated toluenes (110C, 179C, 205C, 223C) for (toluene, benzyl chloride, benzal chloride,
benzotrichloride). A typical graph of temperature variation is shown below - the flat plateau in the middle of the first graph corresponds to TCCA
change.
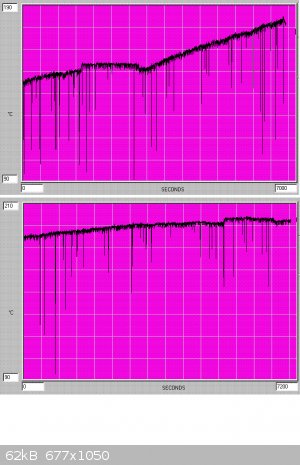
When a b.p. of 220C is reached the reaction products are a quite dark yellow-brown, and continuation of chlorination at this stage will lead to
excessive product decomposition due to a combination of high temperature and little light penetration. At this stage, after 700gms TCCA had been used
in 7+ hours, the reactor was cooled (slight disconnection of all hot joints is recommended immediately) and the product distilled into a clean fresh
reactor flask.

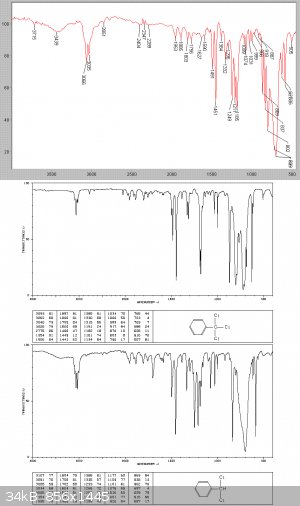
325 grams of a clear heavy-ish liquid were collected at this stage, with 31gms of dark condensation tars left in the still. A Beers law analysis of
the IR graph below, based on the individuating absorptions at 586 and 629 cm-1 showed that this is about a 50/50 mix of the di and tri chlorinated
products.
Benzotrichloride - step 2
According to US patent 3816287 a substantial amount of ring chlorination and tar product formation (32%) accompanies ordinary industrial chlorination
to benzotrichloride. They suggest use of several stages, as well as low reaction temperatures and dilution of Cl2 with inert gas in the final stage to
achieve a product pure to the 0.2% level. They claim to use only a 30% Cl2 excess (in their case actual generated chlorine rather than TCCA based).
I have repeated their procedure, but found the use of inert gas (argon in my case) unnecessary. Indeed reduction of Cl2 flow rate should achieve the
same effect, with the diluting medium provided by the chlorinated toluene vapour pressure. The only disadvantage of this is a lower rate of HCl
efflux, however the reaction is essentially right shifted and a build up of products has little effect on the equilibrium. Hence towards the end I
turned off my Ar flux.
Chlorination, which commenced with the liquid temperature held steady at 170-180C, was accompanied by a dense white fog which the Cl2 flux generated,
and the liquid phase turned a ligh yellow due to Cl2 dissolution. It never however reached a brown colour in this case. Some Cl2 was now evident in
the scrubber and reflux condenser. At 2hrs the temperature was reduced to 150C. At some point the white fog disappeared, and the Cl2 now bubbled in
a clear yellow liquid - this indicates the end of reaction. The reactor was cooled, and the intial charge of 273gms was found to have gained 37gms
weight. The product was now light yellow, and upon exposure to the air on days with humidity > ~50% fumed.

This weight gain is slightly in excess of theoretical and is due to some dissolved Cl2, since some evolution of yellow gas was observed at the intial
stages of redistillation of this product at the beginning of the next procedure. The product was IR analysed, and is seen to be effectively pure
benzotrichloride.
Benzoyl chloride and phthalic anhydride synthesis from phthalic acid
At this stage my aim was to use the pure product from step 2 above in Kyride's procedure (frequently advocated by Sauron) to simultaneously synthesise
benzoyl and phthlyl chlorides. I redistilled the PhCCl3 as Kyride recommended, and halved all the amounts he used in the synthesis.

The 'phthalic anhydride', and zinc chloride were purchased from a reputable distributor.
A key feature of this synthesis is that all apparatus and reagents must be absolutely dry - every gram of H2O present initially will 'destoroy' 5.4gms
of PhCCl3, converting it to the unreactive benzoic acid. The ZnCl2 presents a special problem being hydroscopic, while its Lewis acid properties
(which make it useful as a catalyst here) also mean it can not be well dried of moisture once absorbed. When heated beyond a certain point it
releases HCl rather than H2O forming zinc oxychloride
ZnCl2.x H2O -> (1-x)ZnCl2 + x ZnOHCl + x HCl
It is possible to dry hydrated ZnCl2 in a stream of HCl (or reacting with thionyl chloride), but these methods are laborious in light of the fact that
benzotrichloride will perform the same drying function itself. A key point is that not too much moisture must be present as to consume above a few
percent of the benzotrichloride, else the stoichiometry will be altered. As a consequence 10gms ZnCl2.x H2O were heated till melting (this occured at
240C as opposed to 275C for the pure substance) and subsequently held at 300C for 1/2 hour (zinc chloride sublimes substantially). This resulted in a
loss of 0.5 grams, assuming this to be entirely HCl (the worst case) we have x=0.17 in the above formula.
The commercial phthalic anhydride was dried for 1hr at 120C and then held over CaCl2 overnight. Quite a bit of white sublimed 'fluff' formed during
this procedure, and this together with the odour (which is quite sharp for the anhydride) gave the impression the sunstance was pure. The reagents
are shown in the picture below.

Since the catalyst is little soluble in benzotrichloride, and the stoichiometric anhydride also does fully dissolve, this is a two-phase reaction, and
hence efficient stirring is required. Due to the substantial quantities involved, this was provided by an overhead motor. To reduce product loss
(this was subsequently well justified) a reflux condenser was attached, even though the recation temperature of 120-130C is well below the b.p. of any
of the reactants/products. Being moisture sensitive a CaCl2 tube was used at the exhaust. The complete setup is shown below.

Once heating was turned on, a vigorous reaction with frothing and gas-evolution set in at 70C. Large quantities of gas, which quickly condensed
moisture and completely filled the fumehood with a white fog appeared. A yellow liquid started to condense and drip from the ceiling of the hood -
analysis showed this to be conc. HCl. After 1hr the reaction died down, further stirring produced no visible results. The fan cleared the fumehood.
Surprisingly some undissolved and unmelted flakes of what was now obviously unreacted phthalic acid remained on the wall of the flask even at 130C. I
analysed the suspect anhydride, and comparing the result with the known spectrum of phthalic acid one can see them identical - except for small peaks
of a minority substance, such as at 1851 cm-1. This in fact is a major absorption of phthalic anhydride. Application of Beers law shows the
commecrical anhydride had hydrolised on storage to now be 90% phthalic acid and 10% anhydride - see spectra below.
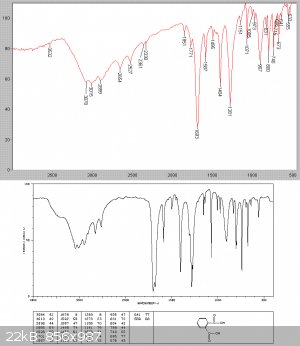
After cooling a solid slurry set it the flask. The reaction mixture was now fractionally distilled at low pressure through a Hempel collumn with 5cm
of glass beads, since vapour assumes so much more volume at reduced pressure and bumping was substantial. 99.9 grams of a clear liquid collected, all
at 78-80C see picture

An IR analysis showed this to be essentially pure benzoyl chloride
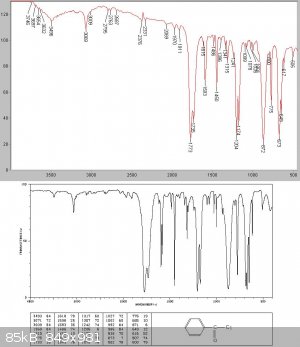
There was (initially surprisingly for me) almost no intermediate fraction, but at about 138C a white solid started to come over. I managed to collect
about 20gms of this before the condenser blocked.
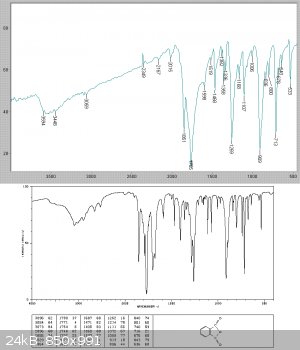
Analysis of the solid showed this to be relatively pure phthalic anhydride. It clearly contained a slight acid chloride impurity as it fumed and
lacrimated the eyes.
Phthalyl chloride synthesis from phthalic anhydride
Having now obtained the phthalic anhydride I thought I initially had, I ground the quantity remaining in the flask (tared brown by PhCCl3
polymerisation byproducts) mixed with the white distilled anhydride, and reacted 60.2 grams of this with 84.1gms of fresh benzotrichloride, adding
3.2gms ZnCl2. 0.17 H2O as catalyst.

The reaction setup was as before, however I used a 250ml flask due to the smaller quantities. There was some HCl evolution at 70C due to the moisture
in the ZnCl2, but this released no more than a fraction of a gram, after which all obvious signs of reaction ceased. After 12hrs of stirring at
120-130C, the reaction vessel was cooled, and this time no solids formed.

Fractional distillation yielded 54.1gms of BzCl which all came over at essentially 82-84C - indicating purity, there was an intermediate fraction of
6.4gms in the 86C-110C temperature range. Almost no product came over untill 132C. The final fraction was collected in the 132C-136C range, with 90%
comming over at the fixed temperature of 136C.

Unlike Kyride no phthalic anhydride crystals condensed from the liquid on cooling, and analysis showed this to be almost pure phthalyl chloride
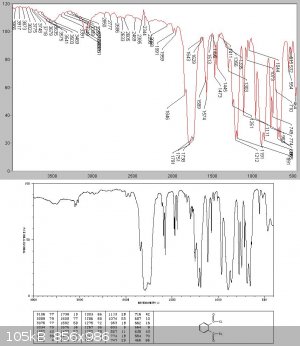
Attempted thionyl chloride synthesis from benzotrichloride and SO2 - no reaction
As mentioned in the introduction this seemed a viable reaction. Some refernces in the literature - patent 3875226 - indicated 58% yields are possible
at temperatures of order 250C and pressures of 30-50 atmospheres, with the SO2 being loaded into the reactor in liquid form below -10C. Obviously
this is a far more complicated route to SOCl2 than I had already had success with and so unpromissing, especially in light of the low yields - which
are to be expected as SOCl2 is unstable at high temperatures. Nonetheless Kyrides obtained 66% yields from phthalyl chloride. Due to the lower b.p.
of the benzotrichloride such a temperature is unsuitable in the presnt case, however the reaction, with dried SO2 was carried out at temperatures up
to 165C and with fast SO2 evolution (I passed a 4-fold excess of SO2, about 1.2 mole through 63.4gms PhCCl3).

At the end of 5 hours no condensate had gathered despite the use of a very efficient condenser. Some small amount of condensation in the connections
was analysed and found to be a roughly equal mix of benzotrichloride, and benzoyl chloride, formed by hydrolysis of residual moisture in the zinc
chloride.
Health issues
The major concerns for this practical are the same as those already described in the benzaldehyde thread, and the same precautions should be taken.
In addition benzoyl chloride is far more lacrymatory than any of the chlorinated toluenes, but has none of the after-effects of benzyl chloride.
Conclusion
Excellelent yields and purities of benzotrichloride, benzoyl chloride, and phthalyl chloride have been obtained in this experiment, starting with
easily obtainable materials. A picture of my blackboard on which I kept track of results during this prac is shown below - as a memory.

[Edited on 10-5-2008 by len1]
[Edit by mod: image restoration]
[Edited on 19-8-2022 by j_sum1]
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Just a couple of corrections as quibbled.
The literature references are properly to JACS not JCS I believe.
Bz as in BzCl is not abbreviation for benzoyl but rather benzyl, BzCl is benzyl chloride, benzoyl chloride is PhC(O)Cl. You can of course redefine
abbreviations arbitrarily but it is confusing.
Phthaloyl chloride is the proper name for the acid chloride (dichloride) of phthalic acid. There is no "phthalyl" chloride that I am aware of.
Anyway I commend you for this article, an excellent addition to your Illustrated Guide series.
I suspect that the failure to replicate the Kyrides prep of SOCl2 was due to the attempt to carryout the reaction at ordinary pressure. Kyrides as you
know used autogenous pressure in an autoclave. I will be performing this reaction in a Parr stirred pressure reactor at some point. This is boring
from a photographic point of view, but is after all the original procedure.
Which was your reputable distributor? Truly anhydrous ZnCl2 is certainly available as is truly anhydrous phthalic anhydride. I would not expect heroic
efforts at drying would be required if one started with freshly opened samples of the correct reagent grades. If necessary, opening and weighing out
in a dry box or dry bag in an atmosphere of Ar or N2 is not such an onerous procedure, is it?
I'd appreciate details on the high pressure Hg illumination source you are using as I just bought medium pressure immersion lamps, power supplies and
special glassware (borosilicate and quartz) for such reactions, 400 W and 600 W. I have seen these quartz-jacket, Hg vapor arc lamps occasionally
described as high pressure, but they are usually described as medium pressure, as opposed to low pressure. I think those relate to watter per inch (or
cm) total emission, the medium pressure lamps usually operate in the 50-100W/cm range. If you are irradiating through the external wall of a
borosilicate flask a lot less than that is reaching the substrate. That is why quartz immersion wells are preferred. But they are costly. For
chlorinations borosilicate immersion wells are all right and allow 100% of the output to be used. The significant wavelength is 365 nm.
Again, a worthy article and a good job continued.
My quibbles are intended to be constructive and hopefully will be taken in that spirit.
[Edited on 10-5-2008 by Sauron]
[Edited on 10-5-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Thanks Sauron. I indeed noticed these articles because of youe efforts on the forum and mention you there.
Thanks, Ill correct the JACS reference.
Kyride's article is titled 'Phthalyl Chloride' and the same spelling is used throughout. I used that spelling in the title out of deference to him,
but the more common 'Phthaloyl' inside the article.
BzCl I believe is correct for PhCOCl, benzyl chloride would be BnCl.
From reading Kyrides account in the paper I mention I could see any references to autoclave or pressurisation - maybe you mean a different paper?
My Hg lamp im afraid is nothing special - a $50 tanning lamp from Hereaus. The ballast is $150 worth, et c'est tous.
The distributor was a local Australian company - not Merck or Aldrich, these overpricers will only sell to my work account. My aim currently is not
to use them Im a bit appaled at what goes around here, but then again should be thankful for an interesting reaction it produced. I wasnt thinking
that though when cleaning up after the 1.5moles HCl released in my hood. Len
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
I buy a lot of Ajax Finechem products from Australia, including ZnCl2. But the ZnCl2 was quite large lumps and needed to be brokem up to use in other
reactions. For sure I would not assume it is anhydrous.
Aldrich sells several anhydrous grades, including powder and -10 mesh beads. These are listed as 99.99% and higher but that is on a trace metals
basis. The specs do not mention any water content, and neither do the specs for ACS Reagent grade which is only 97%.
I'd put it in a vacuum dessicator and pull a good vacuum on it over P2O5 overnight and then I'd melt it in an inert and definitely dry atmosphere.
That's if I had to start with any "suspect" grade like my lumpy Ajax.
The Ajax is fine for most reactions, as is.
Kyrides specifically stated autogenous pressure for the prep of phthaloyl chloride, that means a pressure reactor. Since the conditions for the prep
of SOCl2 from SO2 were over 200 C for a protracted period, that was clearly a pressure reaction as well, you simply can't reflux SO2 - it's just going
to stay absent from the reaction mix. SOCl2 product is also going to all be in vapor phase. Autogenous pressure will do the trick. I will go reread
Kyrides to make sure.
Sorry, I'd forgotten Kyrides spelling.
[Edited on 10-5-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
Excellent work again, Len.
You have demonstrated that one does not have to use phosphorus compounds or thionyl chloride in order to prepare acyl chlorides.
All it takes is chlorine, toluene, and ZnCl2.
If benzoyl chloride is the sole desired product, benzoic acid can be used instead of phthalic acid or -anhydride for reaction with the
benzotrichloride!
The production of acetyl chloride from benzoyl chloride and acetic acid has been documented on the german forum here:
http://www.versuchschemie.de/ptopic,172755.html#172755
The principal finding is that anhydrous zinc chloride seems to strongly catalyse this exchange of chlorine with hydroxy, just as done here with the
benzotrichloride.
Simply mixing benzoyl chloride and acetic acid and heating did not do the job, only after adding a catalytic amount of ZnCl2 was there obtained a
distillate of acetyl chloride.
However, the yield was not very good, and there seemed to be a side reaction, maybe acylation of the aromatic nucleus by the acetyl chloride, also
catalyzed by ZnCl2.
About phthalic acid and -anhydride: Ullmann gives 210- 211°C as the temperature at which phthalic acid eliminates water and turns into the anhydride.
As the boiling point of phthalic anhydride is 295°C, one would have to do this in a round-bottom flask to avoid excessive sublimation, with heated
upper section of the flask to keep the sublimate molten and have it run back into the melt.
Another strategy would be to heat the phthalic acid in a high-boiling hydrocarbon and continuously distill off the water.
Perhaps there are some catalysts that facilitate the anhydride formation, so that maybe toluene could be used as the water carrier, allowing a
dean-stark trap to be used.
About ZnCl2: If this has become wet, simply melting it under exclusion of moisture will cause some hydrolysis to basic zinc chloride.
Anhydrous ZnCl2 can be made by oneself from Zn metal and chlorine in a heated tube, the product resublimates in the cooling zone.
Just like AlCl3, ZnCl2 can be purified by sublimation.
[Edited on 10-5-2008 by garage chemist]
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Thanks garage chemist. Maybe i should try to repeat that AcCl synthesis but with the benzotrichloride - the german patent gives no indication of
yield, but if thats good going thru benzoyl chloride might not be necessary.
You can make BzCl from the trichloride even with just water and a Lewis acid, but the benzoic acid route you mention gives twice the yield based on
the trichloride.
Im interested on th Zn + Cl2 nethod you mention. I previously also searched for this method but in vain. All I could find was Zn and HCl, but that
was at 700C
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
H.C.Brown's "volatile acid chlorides" procedure employinh 1.5-2 equivalents of benzoyl chloride at reflux, with fractionation to strip entrained
reactant/product from the HCl offgassing, works very well, for acetyl chloride and lots of others, and no ZnCl2 is employed. Typical yields 85%-93%.
I would not bet against "Boron" Brown.
Len, it appears my memory was playing tricks on me. Kyrides did not teach use of an autoclave for either the SOCl2 prep or the phthaloyl (phthalyl)
chloride prep. The latter worked at 110-120 C with a 20 g charge of ZnCl2 and in same time (10 hrs) with 2 g. at 200 C.
The SOCl2 procedure was a slow stream of SO2 introduced over 20 hrs (overnight).
So I do not know why you succeeded with the phthaloyl chloride but not the thionyl chloride. Are you sure the SO2 was thoroughly dried?
[Edited on 11-5-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
You might be interested, len1, in the 1937 US patent to I G Farben (famous German chemical industry giant) which covers UV catalyzed chlorination of
toluene to benzotrichloride.
On a bench scale of two hunded g toluene, they started the process at 100 c and noted a temperature rise to 125 C, at which temperature the
temperature was maintained by either adjusting chlorine flow or external cooling. After admitting about 1/2 of theoretical Cl2, the absorptioon of Cl2
slowed and temperature dropped, but was maintained by external heating at 125 C. Total time 1-2 hours and yield almost quantitative.
McBee's group modified I G Farben's process in 1947 in their chlorination of xylenes to hexachloroxylenes, teaching that initial temperature of 70 C
and maximum temperature of 110 C were needed to minimize chlorinoolysis and "burning" in early stages, perhaps the source of your tarry polymers.
I will be using Hg medium pressure 400 W immersion lamp in a Pyrex double walled, cooled immersion tube for this work, initially on a 425 g charge of
m-xylene, which per I G Farben gives a 1200 g yield of very pure crystalline HCMX after cooling. Total chlorination time 4-10 hours depending on rate
of introduction.
I don't need to make benzotrichloride as I buy it and have plenty on hand, but I have not found a source for HCMX except Aldrich Rare Chemicals (I
didn't even ask doe quote) and HCPX, the para isomer, is $9 a gram from Acros.
[Edited on 22-8-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
I couldnt find a Farben patent from 1937 dealing with benzotrichloride, are you sure?
Also the Kyrides procedure I have (the same as posted on the forum) has the SOCl2 from SO2 at 200C not the 100C you mention. This is what makes the
procedure low yielding as SOCl2 is unstable above about 145C
2SOCl2 -> 1/2S2Cl2 + SO2 + 3/2Cl2
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Here's the IGFarben US patent, attached below. A number of the papers I have cite the British and French versions of same patent, numbers are
different but inventor, assigness and subhect matter are same.
You are right, Kyrides ran his reaction at 200 C, but he did get a 66% yield of SOCl2. The dog walked on its hind legs. It is true that a lower
temperature would be better. But all he was doing was demonstrating the reversibility of the reaction forming phthaloyl chloride from SOCl2.
Furthermore in light of much science and technology 50 years later, we know that ZnCl2 as catalyst can be improved upon and that the reaction with SO2
and trichloromethylbenzene and optimal catalyst is best done in liquid phase at ambient temperatures or a little elevated. In fact the US patent to
Dow that you cited, teaches not more than 125 C and 40 atmosphere autogenous pressure. That's just 600 psi, too much for glass but a walk in the park
for steel.
Kyrides' method for chlorinating phthalic anhydride with SOCl2 is only of academic interest. His route from benzotrichloride is much better.
And now we have far better routes to SOCl2.
If I wrote 100 C it was a typo and I will correct it if not too late for Edit.
[Edited on 22-8-2008 by Sauron]
Attachment: US2132361.pdf (185kB)
This file has been downloaded 1502 times
Sic gorgeamus a los subjectatus nunc.
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
I had this patent (its 1938) but it deals only with chlorination of xylenes etc.
The findings regarding latter are pretty much as I wrote above for toluene - stories that as the 3rd hydrogen in the methyl is substituted ring
chlorination and condensation increase markedly are patently untrue
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
You must speed read these things, as you are missing a lot.
The examples describe, in addition to xylene chlorination, chlorination of p-chlorotoluene to p-chlorobenzotrichloride; chlorination of toluene
itself; chlorination of mesitylene and a couple ring halogenated mesitylenes. And so on. Details for the p-chlorotoluene chlorination are applied to
toluene chlorination. That is, 1-2 hours total time, max temperature 125 C, control of temperature in early stage and external heating in latter
stage. That is on the basis of c.200 g toluene extrapolated from the charge of chlorotoluene.
See p 2, left column line 51 for Example 2, the p-chlorotoluene, and line 9 of right column same page for toluene itself, paragraph beginning "In a
similar manner"
Also see the Ind Eng Chem papers I posted along with this patent in the thionyl chloride thread. Those authors teach a reduced max temp for the
xylene chlorination as compared to the IGFarben teachings. And they deal with the excess of chlorine by either recycling it back to the pot after
scrubbing out HCl, or, using it unscrubbed for initial phase of a second pot and charge. In either case chlorine utilization is quite efficient.
The lower temp avoided the tarring andd polymerization losses you seem to have experienced.
To recap, the IGFarben patent teaches chlorinating xylenes at 150 C and toluene at 125 C
The McBee papers teach chlorinating xylenes at not more than 110 C
You chlorinated toluene at 180 C which is 30 C higher than IG Frben for xylene and 70 C higher than McBee (Purdue Univ) for xylene, and 55 C higher
than IGFarben for toluene.
It's something to think about.
As for chlorine economy there's more than one way to skin that cat.
[Edited on 22-8-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
My question doesn't have much to do with the synthesis of benzal chloride (& analogues) but with the cleanup of glassware following the synthesis.
Some time ago I was making benzal chloride by sparging Cl2 into toluene using a fritted glass gas dispersion tube. I abandoned the attempt but
apparently made enough of the benzal chloride (and/or analogues) to make the frit take on a permanent stench. As I recall I tried toluene and acids
at the time but they didn't seem to do much good, ie, the smell was still there. Today I tried Fenton's reagent with fairly good results, ie, most of
the smell is gone.
Has anyone else had this experience? What would be your recommendations?
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Again, I would recommend a base bath soak! 
It gets rid of most of the organic residus, even sulfide smells disappear overnight, tars, stains that seem embedded in the glass, etc. A simple
copious rince with water, and your glassware is perfectly clean.
This also works for old pyrex baking dishes but you have to do that with a
freshly made base bath, to avoid any exposition to metals salts, or various toxic organic products that accumulate. The condensation products from the
ethanol denaturants are easily washed off, a final wash in the dish washer, and wife is happy and thankfull
You can prepare you base bath by adding IPA or EtOH to conc. NaOH/KOH solution (ratios are availble on thsi forum or elsewhere on the net), to be
occasionaly topped off with a little more alcohol to compensate evaporation. I use a 30L plastic tube, half full, with a firm lid. You only need to
change it every 6-12 months depending on how much you use it. It's a good thing to wash the glassware with acetone before. After a certain period, the
base (NaOH or KOH) gradually turns to insoluble carbonates. The mixture quickly turns brown/black afetr preparation, from the condensation of the
alcohol denaturants, but it's not a problem at all.
You need full facial protection, and long thick base-resistant gloves to fish out the glassware.
The bath itself can represent a certain hazard, so always keep it in a corner on the floor, with a tight lid, under a cupboard or similar, far from
the reach of children, animals or incompetents.
I never could go back to usual scrubbing since I've done my first base bath, and I will never praise it more than enough
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
| Quote: |
Again, I would recommend a base bath soak!
|
Thank you Klute - I remember now your previous recommendations. But do you think this would be OK for fritted glass (a porous plug)? The reason I
didn't try an NaOH solution at the time was I was afraid it might ruin the frit porosity.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
I always wash my glass frits (from very fien to rather large porosity) in the base bath, but don't leav them in more than 12-24H max, and rince them
well by suction water through them, then acetone. Works very nice, except for carbon residus where Piranha is the best option IMHO.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
I recently finished the synthesis of benzotrichloride from 100 mL of toluene. The chlorination to this stage took approximately 19 hours. The product
boiled between 201-208 degrees Celsius at 580 mmHg. I used a 60 Watt incandescent light bulb to assist in the chlorination. The redistilled product
has a density of 1.36 g/ mL. I wish to convert all of it to benzoyl chloride by reacting it with benzoic acid. I plan on fusing the benzoic acid
before the synthesis. I do not have any anhydrous zinc chloride. Could anhydrous aluminum chloride be used as the catalyst for this synthesis since I
have some of this compound?
Amateur NMR spectroscopist
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
580mm? Where are you, up in the mountains?
ZnCl2 is what I have used, its a moderate Lewis acid. Will AlCl3 work? - Its a very strong Lewis acid and could promote all sorts of unwanted
reactions, such as Friedel Crafts acylation by the benzoyl chloride of the aromatic ring. Short of trying though I could not say how strongly this
competes with the above reaction.
You are doing some quite complicated synthesis for the home. Why bother? You could do an honours course at university and actually tackle some
problems of current interest as well as get some recognition for your work.
|
|
|
Jor
National Hazard
Posts: 950
Registered: 21-11-2007
Member Is Offline
Mood: No Mood
|
|
Can;t you just react a little zinc with dry HCl or Cl2? You just need a small amount of ZnCl2 right?
Len1, then why did you perform the synthesis at home? Many people do these things at home for fun, for their interest.
Why react is with benzoic acid? You'd better react it with phthalic anhydride (with ZnCl2 cat.) to get phthaloyl chloride and benzoyl chloride
byproduct. this way you produce 2/3 the amount of benzoyl chloride, but have a very valuable, in the case 'byproduct' (wich caneven be used to convert
SO2 to SOCl2).
Benzoyl chloride, personally is something I would buy as it's very cheap and takes a lot of work in this case. And AFAIK you have access to most
chems.
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
My research laboratory is located at 8000 feet, so the boiling points are considerably reduced. I live near the university that I attend, but the
chemistry department is not interested in chemistry research. The facultys idea of research is running one reaction per semester at most. No advanced
classes are offered in organic chemistry. I have more freedom and accessiblity to equipment and chemicals at my home then at the university. Many of
my professors are scared of me because I know so much about chemistry. Our chemistry degree should be a math degree with a chemitry minor, since we
have to take more math classes then chemistry classes. This is one of the main reasons that I conduct these home experiments besides the fact that I
enjoy doing them.
Amateur NMR spectroscopist
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
This must be a small state university in idaho or montana somewhere engaged primarily in teaching with the academics long past doing research. Pity.
I was recently looking over the prospectus for honours students at a local university and thinking - 'if I could turn the clock back twenty years' -
so many interesting chemistry projects in an all research university. The only thing I object to is the authoritarianism university staff impose on
their students (and each other!).
I recall even the ZnCl2 becoming to an extent charred during the hydrolysis due to the Lewis acid catalysed condensation leading to tars etc., this
problem I expect would be much greater with AlCl3. But nearly ZnCl2 (some oxychloride unavoidable) can be made by very slowly drying a solution of Zn
in hydrochloric acid - over HCl vapour in the last stages.
Actually i dont think idaho or montana has any university cities that high. So perhaps colorado or new mexico.
[Edited on 15-12-2009 by len1]
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
ZnCl2 can be made by gassing Zn powder suspended in dry ether with HCl gas. H2 is evolved from the Zn.
Decanting from unreacted Zn and distillation of the ether leaves anhydrous ZnCl2.
Someone at the german forum once distilled acetyl chloride from benzoyl chloride and acetic acid, catalysed by a small amount of ZnCl2. The yield
wasn't very good though.
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
Len1, your are right about the academics not doing research, once they get tenure, they quit for good as far as research goes. Your are close in your
guess about what state I live in, try Utah were everything's immoral or Illegal! I am going to run a microscale test with my aluminum chloride and I
will report back my results. I planning later this week to chlorinate some more toluene to the benzal chloride stage so that I can make some
benzaldehyde.
Amateur NMR spectroscopist
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
Benzoyl chloride can be produced in high yield using aluminum chloride as a catalyst.
150g of the benzotrichloride I synthesized was placed in a 1000 mL 3-neck flask. 94g of benzoic acid was placed in the flask also. The flask was
equipped with a mechanical stirrer, reflux condenser with a calcium chloride tube and the 3rd neck of the flask was stoppered. 5.0g of commercial
anydrous aluminum chloride was added to the mixture. the solution was stirred rapidly in the process. The mixture turned yellow and large amounts of
hydrogen chloride was produced; this was lead into a beaker of water for disposal. The mixture became fluid and was heated with a mantle just below
the reflux temperature for 1.5 hours. The mixture turned to a tan color. The mixture was then distilled, collecting the fraction boiling between 180 C
and 195 C at 580 mmHg. 80% of the mixture distilled at 189 C. The distilled product weighed 165g for a percentage yield of 76%. The liquid fumed in
air and had a very irritating smell. I did not taken an infrared spectrum because I did not want to expose the instrument to the HCl fumes.
[Edited on 11-1-2010 by benzylchloride1]
Amateur NMR spectroscopist
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Your benzoyl chloride is impure. Benzotrichloride fumes on humid days, benzoyl chloride does not fume at all - I havent tried it in 100% humidity,
but then you are not in the tropics. I certainly did not measure its IR spectrum while exposing the instrument to acid fumes from its hydrolysis.
It is more irritating to the eyes than the nose (it has a benzaldehyde cum fruity smell). So I am not convinced by this AlCl3 catalysis.
[Edited on 11-1-2010 by len1]
|
|
|
| Pages:
1
2 |
|








