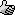| Pages:
1
2
3
4
5 |
Sandmeyer
National Hazard

Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
It's best to explain in pictures, following picture explains why I would call it aza-Cope and not aza-Claisen (sorry for the bad graphics, it is time
to sleep):

I mentioned the presence of a carbonyl in product 1,5-diene as what distinguishes the aza-Claisen from aza-Cope, but hydrolysis of imine to a carbonyl
is a separate step and not part of the pericyclic reaction. It follows that Claisen proceeds via O-allyl-enol species while aza-Claisen via analogous
N-allyl-enamine species and so, logically, the reaction Arrhenius mentiones can not be considered as aza-Claisen since it does not proceed through a
nitrogen analogue of allyl vinyl ether (Claisen), his example fits aza-Cope rearrangement more... All in my opinion of course... 
[Edited on 17-8-2010 by Sandmeyer]
|
|
|
Polverone
Now celebrating 21 years of madness
|
Thread Topped
18-8-2010 at 07:42 |
franklyn
International Hazard
Posts: 3026
Registered: 30-5-2006
Location: Da Big Apple
Member Is Offline
Mood: No Mood
|
|
A Guidebook to Mechanism in Organic Chemistry 6 ed
recommended by DJF90 - http://www.sciencemadness.org/talk/viewthread.php?tid=14305#...
http://rapidshare.com/files/126713899/A_Guidebook_to_Mechani...
Arrow-Pushing in Organic Chemistry
recommended by Ozone - http://www.sciencemadness.org/talk/viewthread.php?tid=14305#...
http://ifile.it/jugotx6/9780470171103.rar
Molecular Electronic Structure Theory Part 1 - Helgaker T, Jorgensen P, Olsen J.
This is entirely steeped in high level mathematical descriptions more suited to physics ( I don't understand it either )
http://rapidshare.com/files/128521981/Molecular_Electronic-S...
or instead
http://rapidshare.com/files/128523134/Molecular_Electronic-S...
.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
No, no, Sandmeyer. That is not what I had in mind. I was comparing the pericyclic step of the transformation directly to the Claisen rearrangement in
a strictly isosteric fashion (N-Boc instead of O and C=N instead of C=C). See the first equation below.
The paper is now kindly made available in the Wanted references thread by Solo. The authors are of the opinion that the protonation occurs
before Boc removal on the basis that equivalent non-Boc hydrazones give poorer conversion (see the relevant part of the article on the right). Seems
plausible, however this also means comparing the sigmatropic rearrangement to Claisen's can not be correct, because this type of rearrangement can
only be catalysed by the protonation of the other nitrogen (the one initially Boc protected). The catalytic effect of the acid by protonation of the
"imine's" nitrogen (which is the only protonable one prior to Boc removal), indicates the electron flow is most likely the opposite to the one in the
Claisen-type of rearrangements (see the third equation bellow). So it can not be a Claisen, but it does not have that much to do with a Cope either,
because Cope does not involve protonation or heteroatoms in that position. Anyway, the authors avoid messing up with such details and do not compare
the pericyclic component of this one-pot tandem transformation neither with the Cope nor the Claisen rearrangement, or any other named reaction, but
consider it as a [3,3]-sigmatropic rearrangement of its own type. So, perhaps it was Arrhenius' idea to call it a Cope-like rearrangement? Or perhaps
they call it that way in one of the two preceding papers that I did not yet check. Or maybe it was Stevens who 30 years ago reported the sigmatropic
rearrangement of allylhydrazones who compared it to Cope. Better not call it names anyway. 

…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
cheeseandbaloney
Harmless
Posts: 21
Registered: 4-4-2008
Member Is Offline
Mood: No Mood
|
|
great discussion! love this shit.
un0me2
Is there a '3d' mode for Symyx Draw? I do like it better than chemsketch, after the adjustment period.
Arrhenius
I forgot to say, checkmate on reaction #4. I'm still steadily learning new important reactions though for expanding the 'moves' I could make. Any more
you think I should tackle, be my guest. One of the most important 'moves' right now IMO is the reformation of the C=O bond. I've noticed it can be the
driving force for a lot of reactions. I remember reading this is because it's very thermodynamically stable. It seems once you find 'patterns' like
this and build on them one can start to become more comfortable in synthetic organic chem.
Ozone
I ended up buying the 2nd book you recommended. Still have a couple to go through at the moment.
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
My advice is try to move beyond Functional Group Interconversions (FGIs). It
takes a sharp memory to write mechanisms for functional group dancing. However, it takes considerable skill to rationalize rearrangements, especially
sigmatropy, etc. That being said, there are certain FGIs with stereochemical outcome that absolutely demand your understanding the mechanism. Also,
given the abundance of transition metal chemistry in organic synthesis, I would suggest you take a look into these mechanisms. I would also recommend
you consider purchasing one of KC Nicolaou's books "Classics in Total Synthesis" - I have the first edition and it's pretty good. The strategy
employed in these highlighted total syntheses is typically quite fantastic. Anyone up to try working through a few more problems?
I've tried to hit on a few practical reasons that make mechanisms and associated transition states important 1.) Rationalizing FGI
outcome 2.) Multi-step mechanisms 3.) organometallic mechanisms 4.) highly strategic bond formation
(see anything by E.J. Corey & co. among others).

|
|
|
Sandmeyer
National Hazard
Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
Quote: Originally posted by Nicodem  | | No, no, Sandmeyer. That is not what I had in mind. I was comparing the pericyclic step of the transformation directly to the Claisen rearrangement in
a strictly isosteric fashion (N-Boc instead of O and C=N instead of C=C). See the first equation below. |
I see, however, it is an incorrect way of reasoning; 1.) since the first equation is messed up, logically LUMO should reside on the C=N carbon, 2.)
since Arrhanius explicitly provided benzaldehyde as example of carbonyl component and not 2-phenylacetaldehyde (enol ether of). As we know,
benzaldehyde has no alpha hydrogens, consequently it can not be used as a direct carbonyl precursor for Claisen rearrangement of any kind, only for
Cope -- as I've depicted above.
| Quote: Originally posted by Nicodem | | The catalytic effect of the acid by protonation of the "imine's" nitrogen (which is the only protonable one prior to Boc removal), indicates the
electron flow is most likely the opposite to the one in the Claisen-type of rearrangements (see the third equation bellow). So it can not be a
Claisen, but it does not have that much to do with a Cope either, because Cope does not involve protonation or heteroatoms in that position.
|
Well, that's not the case, aza-Cope is catalyzed by protonation of imine nitrogen, logically, since the LUMO energy of iminium
species is further lowered compared to that of the corresponding imine. We all agree that Cope and Claisen rearrangements are [3,3]-sigmatropic
rearrangements, however, the reaction Arrhenius refers to can not rationally be considered a Claisen rearrangement since benzaldehyde can not
enolize/enaminaze, I don't think this is such a far out idea, I think it is quite basic and obvious but for some reason you are avoiding it. You did compare the direction of the electron flow in the Arrhenius example with
that of Claisen rearrangement, but more importantly you didn't mention that the electron flow in the Arrhenius example is fully consistent with that
of aza-Cope rearrangement, i.e C=N carbon - LUMO, alkene - HOMO.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Sandmeyer |
I see, however, it is an incorrect way of reasoning; 1.) since the first equation is messed up, logically LUMO should reside on the C=N carbon, 2.)
since Arrhanius explicitly provided benzaldehyde as example of carbonyl component and not 2-phenylacetaldehyde (enol ether of). As we know,
benzaldehyde has no alpha hydrogens, consequently it can not be used as a direct carbonyl precursor for Claisen rearrangement of any kind, only for
Cope -- as I've depicted above. |
Ah yes. The flow of electrons is likely wrong in the first equation. I always have troubles with deciding which Pi-system interacts with its LUMO and
which one with its HOMO. That orbital stuff was never appropriately thought to me (unfortunately we had very old professors). I never trust much my
intuition on this and whenever I run on similar problem I go asking a friend to calculate energies with Gaussian (even though I have serious troubles
trusting computational chemistry, but obviously it is more accurate than my intuition). I guess this is one of those things I was trying to explain in
one previous post, that one should learn these things while young. It is too late trying to do so later when you work in the lab and have children at
home.
Your second point is however something I can't really integrate in the discussion. The O-allylation of aldehydes does not constitute as part of the
Claisen rearrangement, so neither can the formation of allylhydrazones be considered as part of the aza-Cope rearrangement. If you compare two
reactions, the comparison simply must be isosteric - you can't base the comparison on the synthesis of the starting compound (named reactions are
defined by the mechanism, though it sometimes turns out that more than one mechanism operates).
| Quote: | | You did compare the direction of the electron flow in the Arrhenius example with that of Claisen rearrangement, but more importantly you didn't
mention that the electron flow in the Arrhenius example is fully consistent with that of aza-Cope rearrangement, i.e C=N carbon - LUMO, alkene - HOMO.
|
I only compared it to Claisen because that is what it appeared like before I had the article in hands. I soon gave up on that comparison, but truly
can not call that an aza-Cope rearrangement either, though I agree that that the electron flow is consistent. How about not calling it anything but a
[3,3]-sigmatropic rearrangements? The names of named reactions are often used confusingly in the literature. There is an inflation of names and an
inflation of their use. You think the use of the name aza-Cope is consistent? Check this:
3-Aza-Cope Rearrangement of Quaternary N-Allyl Enammonium Salts. Stereospecific 1,3 Allyl Migration from Nitrogen to Carbon on a Tricyclic
Template

Is that an aza-Cope or an aza-Claisen? To tell you the truth, now that you got me so confused, I can't be sure any more (even though a few days ago I
would say Claisen before even blinking with an eye). So, what you call aza-Cope would be 2-aza-Cope and what I would call aza-Claisen would be
3-aza-Cope. But that mean it could be the opposite as well! Some might call your aza-Cope an 2-aza-Claisen. Yet, how about HOMO & LUMO roles, and
the flow of electrons? I think that until IUPAC defines the proper use of heteroatomic prefixes in named reactions, one should avoid their use and
stick to naming mechanisms utmost (but only when enough evidence is obtained).
[Edited on 20/8/2010 by Nicodem]
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Apparently, and to my surprise, it has become an incurable habit to consider any hetero-Cope or "other than 3-oxa"-Claisen rearrangements as
synonymous. At least that is what the author of the review on aza-Claisen in Top. Curr. Chem. 244 (2005) 149–213 says it's been going on in
the literature.
| Quote: | Introduction
3,3-Sigmatropic rearrangements are defined as uncatalyzed processes to migrate
a sigma bond of two connected allyl systems from position 1 to position 3.That
means both allyl systems suffer from an allyl inversion. Though described for
the first time in 1940, the Cope rearrangement can be considered as the basic
type of such a process, since C–C bonds only are reorganized during the course
of the reaction [1].More than two decades earlier, in 1912, L. Claisen first described
the rearrangement of aromatic allyl vinyl ethers to generate o-allyl
phenols [2]. This so-called Claisen rearrangement is characterized by the replacement
of the C3 carbon of the rearrangement system against a heteroatom
X. The basic Claisen rearrangement bears X=O; consequently, such a process can
be termed as a 3-oxa Cope rearrangement. Analyzing the literature, rearrangement
systems displaying other heteroatoms X in position 3 can be found as hetero
Claisen and 3-hetero Cope rearrangements.Focussing on systems with X=N,
names such as aza- and amino-Claisen as well as 3-aza-Cope rearrangement
occur in the literature.Furthermore, the term aza/amino Claisen rearrangement
is widely used for nitrogen introduction processes rearranging 1-aza-3-oxy-
Cope systems (imidates) to generate carbamates. Finally, the Fischer indole
synthesis represents a special type of aza-Claisen rearrangement incorporating
two N atoms in the 3 and 4 positions of the rearrangement system.
Intending to set a firm basis concerning the notion of the sigmatropic
rearrangements, the following review will use the term Claisen rearrangement
for 3,3-sigmatropic core systems incorporating a heteroatom X in position 3, i.e.,
an aza-Claisen-type rearrangement is characterized by X=nitrogen (Fig. 1). |
So, regardless of the electron flow, structural peculiarities, etc., we can call the example at hands either as 2,3-diaza-Cope or 2,3-diaza-Claisen
rearrangement. It makes no difference even if we see a difference. I just hate (stupidly) named reactions!

…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Sandmeyer
National Hazard
Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
| Quote: Originally posted by Nicodem |
Ah yes. The flow of electrons is likely wrong in the first equation. I always have troubles with deciding which Pi-system interacts with its LUMO and
which one with its HOMO. That orbital stuff was never appropriately thought to me (unfortunately we had very old professors). I never trust much my
intuition on this and whenever I run on similar problem I go asking a friend to calculate energies with Gaussian (even though I have serious troubles
trusting computational chemistry, but obviously it is more accurate than my intuition). I guess this is one of those things I was trying to explain in
one previous post, that one should learn these things while young. It is too late trying to do so later when you work in the lab and have children at
home. |
No need for Gaussian, to figure out the direction of electron flow in this problem it is enough to know that enamines are nucleophilic on beta carbon
while N-Boc hydrazones, in analogy to imines and contrast to enamines, are electrophilic. 
EDIT: The review Nicodem refers to, and that settled the matter of naming the reaction, seems like an interesting read, can someone please post it,
thanks! 
[Edited on 22-8-2010 by Sandmeyer]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Sandmeyer | No need for Gaussian, to figure out the direction of electron flow in this problem it is enough to know that enamines are nucleophilic on beta carbon
while N-Boc hydrazones, in analogy to imines and contrast to enamines, are electrophilic.
|
I'm not so confident when it comes with less familiar systems like this hydrazone one. Also, it is not only the double bond systems that need
comparison, but since it is about [3,3]-sigmatropic rearrangements the whole allylic and "heteroallylic" (enaminic, enolic, etc.) moiety needs to be
considered to find what is the energy difference between the HOMO & LUMO. Also, the electron flow will determine what formal charge needs to be
considered in the calculation / intuitive evaluation. It is not always trivial to comprehend which moiety reacts with its HOMO and which one with the
LUMO (as the combination that has the lowest energy difference might not be obvious at first glance). Furthermore, things change with acid catalysis.
So you end up with having to consider a bunch of orbital combinations and find out which HOMO-LUMO interaction is most favourable just to realize if
the reaction is viable at all, possibly at any "normal" temperatures, and if protonation or Lewis acid coordination catalyses it. I wish someone with
more knowledge of pericyclic reactions could describe a few personal tricks for intuitive evaluation of the reactions.
For example, what would you expect the order is in HOMO and LUMO energy of systems like RCH=N-NBoc-, RCH=CH-NBoc-, RCH2=CH-NMe-,
RCH=CH-N<sup>+</sup>Me<sub>2</sub>-, RCH=N<sup>+</sup>H-NBoc-, RCH=N-O-, RCH=N<sup>+</sup>H-O-, etc.?
I'm afraid I don't know. If the question was which of these fragments when combined with an allyl group would most likely rearrange at the lowest
temperature, I could not answer easily without resorting to Gaussian or Schrödinger-Jaguar. And even then, interpreting the results leaves much too
much uncertainties.
PS: Find the review attached. (BTW, nearly the whole seriez was made available in PDF if you know where to search)
Attachment: Recent Advances in Charge-Accelerated Aza-Claisen Rearrangements.pdf (1MB)
This file has been downloaded 1856 times
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Sandmeyer
National Hazard
Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
| Quote: Originally posted by Nicodem |
I could not answer easily without resorting to Gaussian or Schrödinger-Jaguar. And even then, interpreting the results leaves much too much
uncertainties. |
Resorting to... Schrödinger-Jaguar huh? Is that a beer?
|
|
|
cheeseandbaloney
Harmless
Posts: 21
Registered: 4-4-2008
Member Is Offline
Mood: No Mood
|
|
Hey, another quick mechanistic question! Was flipping through some of the chapters in one of my orgo books ferking around with some of the problems.
Came across what seemed like an easy one, but was an answer I didn't expect. What would you fellow chemists consider the major product of this
reaction?

|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
The major products would be cyclohexanone and methyl iodide. Mechanistically, the vinyl ether is protonated on *carbon* with formation of an
oxocarbenium ion, then iodide attacks at the methyl carbon of the methyl vinyl ether. Hydrogen iodide mediate cleavage of aryl methyl ethers should
proceed through the same mechanism. I do believe it can cleave other ethers, but this is definitely a brutally harsh reagent. Trimethylsilyl iodide
(TMSI) is often employed to cleave ethers, especially methyl ethers, as a gentler alternative.
Cheers & keep reading!

[Edited on 13-10-2010 by Arrhenius]
|
|
|
cheeseandbaloney
Harmless
Posts: 21
Registered: 4-4-2008
Member Is Offline
Mood: No Mood
|
|
yo Arrhenius! Why does the iodide specifically attack the methyl group and not the carbonyl carbon?
edit: wait, does it haff to do with the stability of products, or enthalpy?
[Edited on 10/14/2010 by cheeseandbaloney]
|
|
|
Ebao-lu
Unregistered
Posts: N/A
Registered: N/A
Member Is Offline
|
|
just a short copypaste from https://www.sciencemadness.org/whisper/viewthread.php?tid=14...
| Quote: |
O-desmethyl tramadol is prepared by treating tramadol as a free base under O-demethylating reaction conditions, e.g., reacting it with a strong base
such as NaOH or KOH, thiophenol and diethylene glycol (DEG) with heating to reflux (Wildes, et al., J. Org. Chem., 1971, 36, 721)
|
What is the mechanism of this kind demethylation? I'm not able to view the original paper unfortunately
My propasal is that it is simply a necleophilic catalysis by thiolate, which is a stronger nucleophile then hydroxide. And at the same time, it is a
better leaving group then alkoxide. So it is Ar-OMe + -SR =intermediate=> Ar-SR + OMe-
Ar-SR + 2OH- =intemrediate=> Ar-O- + SR- + H2O
But that intermediate is very unstable, thats why high temperature is required.
Is it correct ?
[Edited on 15-10-2010 by Ebao-lu]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by cheeseandbaloney | yo Arrhenius! Why does the iodide specifically attack the methyl group and not the carbonyl carbon?
edit: wait, does it haff to do with the stability of products, or enthalpy? |
Addition of HI (or HCl, HBr...) on the carbonyl group is reversible with the equilibrium very highly in favour of the carbonyl rather than
gem-halohydrins. The more likely further reaction of cyclohexanone under very acidic conditions is the acid catalysed aldol condensation, but this
should be slower than the vinyl ether cleavage, so it should not represent a problem under properly chosen conditions.
If aqueous HI is used for the demethylation then the competing reaction is the addition of H2O on the intermediate O-methylcyclohexanonium
carbocation. This is a normal hydrolysis of enol ethers which however leads to the same product (cyclohexanone) and methanol. If HI is concentrated
enough, methanol ends up forming methyl iodide via SN2). Thus you have an interesting situation where the end products are the same regardless of the
mechanistic pathway. This is actually a common situation where two or more mechanistic routes lead to identical products from the same substrate and
the (major) pathway can only be demonstrated by observing (spectroscopically, chromatographically, etc.) or catching the intermediate (in this case
methanol in the second pathway, but absent in the first).
Like most demethylations (if not all) it is just a normal SN2 substitution. Similarly as the demethylation mentioned the two posts above yours.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Ebao-lu
Unregistered
Posts: N/A
Registered: N/A
Member Is Offline
|
|
Btw, Nicodem and Arrhenius, the rearrangement you are talking about could be also semi-homolytic, like in Fisher's indole synthesis and
Hoffman-Loeffler reaction. That means, any arrows should be avoided in the mechanism, because arrow is an electron pair, not a single electron.
Usually such rearrangement mechanisms, if i remember correctly, are drawn like dotted line if i'm not mistaken, and BOC-protected aminogroup is just
an analogue of protonated nitrogen in ability to form cathionradicals like RNH2(+*) or
BOCNH* <-> t-BuO-C(O-)=NH(+*) <-> t-BuO-C(O*)=NH
[Edited on 26-10-2010 by Ebao-lu]
[Edited on 26-10-2010 by Ebao-lu]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Ebao-lu | | Btw, Nicodem and Arrhenius, the rearrangement you are talking about could be also semi-homolytic, like in Fisher's indole synthesis and
Hoffman-Loeffler reaction. That means, any arrows should be avoided in the mechanism, because arrow is an electron pair, not a single electron.
|
The crucial step in the Fisher's indole synthesis is a pericyclic reaction, namely a [3,3]-sigmatropic rearrangement, thus there is a circular flow of
electrons (as pairs). Normal arrows are therefore used to depict what goes on. I never heard about articles claiming it involves a homolytic cleavage
of bonds.
The Hofmann-Löffler reaction does involve a homolytic bond cleavage, but I see no similarities with the above [3,3]-sigmatropic rearrangements.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Ebao-lu
Unregistered
Posts: N/A
Registered: N/A
Member Is Offline
|
|
i'm sorry. the rearrangement you are talking about is not that kind, as there is no N-N bond cleavege at all! did not read it carefully first
| Quote: | | I never heard about articles claiming it involves a homolytic cleavage of bonds |
here is one http://www.springerlink.com/content/x0h4047554788212/fulltex...
I have also seen most articles that claimed an arrow-drawn mechanism of fisher indole synthesis, that is maybe due to simplicity
Indeed, i think there is no _ideal_ synchronous sigmatropic rearrangement. Some are bit heterolytic(even having extremely short living ion
intermediates and such), some bit homolytic.. depending on what is more beneficial.
Otherwise how a charge-attraction product formation in some diels alder reactions could be explained? Sometimes it is not thermodinamically favored
over others, so that is definitely due to kinetics, or in other words activation energy difference. The latter may result from higher energy of
transition state in case of unfavored product and lower in case of proper (charge to charge) orientation. If we draw them both then it is clear that
it is mostly because of charge interaction, and sometimes there is no substantial repulsion in case of non proper product, there is just an attraction
in proper. But those charges are derived from pi-system, that means it is not an ideal 4+2 reaction!
[Edited on 26-10-2010 by Ebao-lu]
[Edited on 26-10-2010 by Ebao-lu]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Ebao-lu |
| Quote: | | I never heard about articles claiming it involves a homolytic cleavage of bonds |
here is one http://www.springerlink.com/content/x0h4047554788212/fulltex...
I have also seen most articles that claimed an arrow-drawn mechanism of fisher indole synthesis, that is maybe due to simplicity
Indeed, i think there is no _ideal_ synchronous sigmatropic rearrangement. Some are bit heterolytic(even having extremely short living ion
intermediates and such), some bit homolytic.. depending on what is more beneficial. |
That is an interesting paper, but it does not demonstrate the mechanism of the Fischer indolization involves a homolytic bond cleavage. The N-N bond
is weak and easily homolyticaly cleaved and thus it is not particularly surprising they got an ESR evidence for the N-N cleavage under the reaction
conditions. This however does not demonstrate this is part of the reaction pathway. Aditional evidence must be presented, for example, an experiment
where spin traps or radical inhibitors inhibit the conversion rate to the indole. Or that the Fischer indolization can be driven photochemically
(though this would only demonstrate the feasibility of the homolytic pathway, but not also that this is the mechanism in the acid catalysed reaction).
Though interested, I currently don't have the time to follow the references cited in that paper, but you are welcome to research this topic and report
back with a short review.
| Quote: | | Otherwise how a charge-attraction product formation in some diels alder reactions could be explained? Sometimes it is not thermodinamically favored
over others, so that is definitely due to kinetics, or in other words activation energy difference. The latter may result from higher energy of
transition state in case of unfavored product and lower in case of proper (charge to charge) orientation. If we draw them both then it is clear that
it is mostly because of charge interaction, and sometimes there is no substantial repulsion in case of non proper product, there is just an attraction
in proper. But those charges are derived from pi-system, that means it is not an ideal 4+2 reaction! |
The Diels-Alder reaction mechanism, as well as its regio- and stereoselectivity, is fully and thoroughly explained on the basis of orbital theory. No
need to call for any other more exotic and off the trail explanation. Mechanistic hypotheses first need to fulfil the Occam's razor criteria. If they
don't, the chances they are wrong or incomplete increase.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Ebao-lu
Unregistered
Posts: N/A
Registered: N/A
Member Is Offline
|
|
Maybe you are right as there is no sufficient evidence that N-N bond homolytic cleavage is part of a reaction pathway, but i don't mean that it is not
3+3 reaction, of course it is. I mean, that in this reaction's transition state there is practically no polarization of N-N bond, so it is cleaved
homolytically with immediate formation of new C-C bond
I think computatuinal techniques can give answer wether there is polarization of N-N bond in fischer's indole synth transition state or not. Other
methods like radical traps etc may not work, because recombination of radicals could be a best trap, especially if they are close or even
synchronously formed and recombinated like in a 3+3 reaction. Spin complex formation like in those article could be just to make them bit more stable,
live for a while or just be formed without synchronous recombination. Another possible method could be measuring the rate of indole formation and
compare it with amplitude of those signals, in different conditions like temperature, catalyst etc. If they are in high correlation, that should mean
it is a part of reaction pathway.
As for articles, i have not read them yet. Btw those guys are having another paper that i am trying to comprehend now where they claim partially
intermolecular mechanism of Fischer http://www.springerlink.com/content/g52q4n254hg33r76/ . The nature of N-N cleavage seems to be debated before according to this article, but i
think not enough. There should be, or maybe there are, articles that gave a definite answer on that question. Did not read cited refferences yet,
besides some need access, which i don't possess.
My point still is, that synchronous rearangements([3+3] [4+2] etc) can be two types - homolytic and heterolytic (or better to say, polar/ non polar)
ones. I dont say that N-N bond in fisher's indole synthesis is cleaved _before_ formation of C-C, both processes are synchronous. But the nature of
this cleavage is homolytical, so there is no polarization in transition state. Drawing arrows in case of any rearrangements was always associated with
polarization of atoms in transition state according to my understanding, but is there any polarization in transition state indeed? That would depend
on substrate. Some substrates would develop substantial polarization in transition state(like in heteroDA reaction), some will not(like in non polar,
symmetrical substrates). But nethertheless arrows are always used to depict mechanism of both polar and non polar transition states, and i think that
is not correct(for non polar ones), so that is mostly for simplicity. I may be wrong, but i dont understans what else can arrows mean but the
direction of electron pairs flow in transition state, and that means polarization
Maybe, theories that are accepted today can give their own explanation of why protonation or a lewis acid can promote fischer indole formation, but
mine would always be the analogy with N-N homolytical bond cleavage, that is eased after protonation because of increased stability of protonated
nitrogen radicals. And in principle i suppose it is quite the same as in accepted theories - the difference is that i don't need computer to calculate
all those electronic orbitals in order to predict what would happen, i just use an approach of easing up N-N bond homolytic cleavage, since nitrogen
radicals become more stable if protonated, and maybe also some impact of electrostatic repulsion between positive charged nitrogen atoms.
I wonder, what the computational model would say concerning this difference between protonated and not protonated hydrazone reactivity towards a
sigmatropic rearrangement, and difference in activation energies between sigmatropic route and homolytic dissociation of diprotonated hydrazone. It
should be quite a simple model for computation. It is quite possible, that activation energy for a sigmatropic rearrangement in this case may be even
higher then homolytic dissociation, because in case of sigmatropic rearrangement's transition state all the atoms are changing their location and that
should results in increasing activation energy, while in homolytic cleavage much less number of atoms are relocating in transition state. This may
also elucidate the mechanism of Fisher indole synth
Maybe all i wrote above in concern of HOMO-LUMO and other related today's accepted theories sounds dumb, but my understanding helps me to understand
mechanisms and i'm satisfied. Most physical or quantum chemists i knew were not very good in organic chemistry, their perfect knowledge of HOMO-LUMO
and other related things(or just book illustration of that) did not allow them to predict outcome of reaction. So apparently, some simplification of
classic theories is allowed.
If there is example where my simplified understanding would not allow to understand mechanism or products formed, i will definitely change my mind and
avoid using it in future.
| Quote: |
The Diels-Alder reaction mechanism, as well as its regio- and stereoselectivity, is fully and thoroughly explained on the basis of orbital theory. No
need to call for any other more exotic and off the trail explanation. |
I am sure, this thorough explanation was given mostly for the simplest cases of D-A. More complicated D-A reaction outcomes are usually explained in a
way similar to this, including charges: http://138.253.125.24/~ng/suzanna/Cycloaddition2.html .And that is the thing i was talking about in the
beginning, about not ideal sigmatropic reactions. Perhaps that is just illustration for students for simplicity, and there are some more thorough
explanations, i never seen them probably because the same reason - being satisfied with existing ones, and not good understanding of psi-functions and
all that kind. I may be wrong btw..
| Quote: | | Mechanistic hypotheses first need to fulfil the Occam's razor criteria. If they don't, the chances they are wrong or incomplete increase.
|
Thanks, i prefer using Einstein's razor! (or Einstein's interpretation of Occam's razor, not sure) that says "Make everything as simple as possible,
but not simpler."
I think i used it  ->. There was no sophistication in my D-A explanation, it was just simplification. Most terms i used are frequently used
in organic chemistry like "activation energy" "charge attraction" (better interaction), "transition state" etc. I did not say that DA mechanism does
not involve orbital interactions(as all reactions, not only DA involve it), i just neglected that as not critical in this particular case for
understanding the regioselectivity and changed it to charge explanation, which does not violate or conflict with any existing rules or accepted
mechanism, being just a simplification of that also in order not to draw orbitals on this board. I was sure you will also not recall about orbitals.
But if you did - feel free to explain that my explanation does not fall under Occam's razor(if you want) - not to debate with you, just for you to
convince me that my explanation is really a sophistication, or over simplification of existing facts and thus may become wrong(i agree that it is
incomplete btw, but it was not a new mechanism proposal, just a simplifacation of existing one).. And i'll change my mind immediately if you do
that, Nicodem ->. There was no sophistication in my D-A explanation, it was just simplification. Most terms i used are frequently used
in organic chemistry like "activation energy" "charge attraction" (better interaction), "transition state" etc. I did not say that DA mechanism does
not involve orbital interactions(as all reactions, not only DA involve it), i just neglected that as not critical in this particular case for
understanding the regioselectivity and changed it to charge explanation, which does not violate or conflict with any existing rules or accepted
mechanism, being just a simplification of that also in order not to draw orbitals on this board. I was sure you will also not recall about orbitals.
But if you did - feel free to explain that my explanation does not fall under Occam's razor(if you want) - not to debate with you, just for you to
convince me that my explanation is really a sophistication, or over simplification of existing facts and thus may become wrong(i agree that it is
incomplete btw, but it was not a new mechanism proposal, just a simplifacation of existing one).. And i'll change my mind immediately if you do
that, Nicodem
[Edited on 29-10-2010 by Ebao-lu]
|
|
|
Copenhagen
Harmless
Posts: 13
Registered: 18-6-2010
Member Is Offline
Mood: Lattice vacancies
|
|
From personal experience as a beginning organic student I found Arrow Pushing by Levy to be the most helpful to me. It was the easiest to read. It
helped my mechanistic intuition immensely. I was able to cover up the products on a lot of the key reactions and correctly predict them by
understanding the fundamentals that Levy teaches.
I also own the Art of Writing Reasonable Reaction Mechanisms and I agree that it is an awesome book on teaching understanding the mechanism. It
includes everything that Arrow Pushing does not. For example it discusses the differences between various electrophiles and nucleophiles and
considering the solvent conditions. "snowballs chance in hell" comes to mind. You won't make the mistake of drawing a hydroxide leaving group in an
acidic medium for example.
Solvent conditions are very important and are not lucidly or adequately explained in any of the course textbooks that I have read. You memorize that
SN2 goes with aprotic and SN1 goes with protic but you don't really learn why...
Arrow Pushing makes it easy to understand that a strong base nucleophile (electron dense) will be shielded by a protic solvent because the positively
charged protons of the solvent will be attracted to negative-electron dense nucleophile and will be less able to react. An aprotic solvent won't
produce such a problem for the incoming nucleophiles. Little things like this are so important in learning mechanisms.
The other books I have found of greater benefit after completing both semesters of organic as they tend to be go in to a lot of detail. Electron Flow
in Organic Chemistry is also a good one but I find Arrow Pushing more to the point.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Ebao-lu | | My point still is, that synchronous rearangements([3+3] [4+2] etc) can be two types - homolytic and heterolytic (or better to say, polar/ non polar)
ones. I dont say that N-N bond in fisher's indole synthesis is cleaved _before_ formation of C-C, both processes are synchronous. But the nature of
this cleavage is homolytical, so there is no polarization in transition state. |
I don't have time to go replying to your post point by point, but will only point out that the most important aspect of pericyclic mechanisms is in
that the bond formation and breaking occurs in a concerted manner. This allows a much lower activation energy to reach the transition state than any
other mechanisms, such as polar or radical ones, could ever allow.
Let me explain you with an of topic example. If you go to a luna park and observe how a Ferris wheel works, you can note that even though it is able
to lift tons of people, it only works on a relatively weak electromotor (requires little activation energy to turn around). That is because the force
to lift (break bonds) is compensated with the force given from the fall on the other side of the wheel (bond formation). This is because both sides of
the wheel (the one going up and the one going down) are connected trough the rotating axis (concerted mechanism). Now if you would have to lift all
those people up to the same height with a lift, give them time to see the view from their transition state perspective, and then take them down with
the same lift, the process would not be concerted any more and it would require much more energy (full lift and wasted fall). This would be so even if
comparing with a unbalanced, unevenly occupied Ferris wheel (not fully synchronous).
I'm specifically comparing pericyclic reactions with a wheel because geometry plays a crucial role - the transition state must be a cyclic structure
so that the bonds can easily form and break in a concerted manner.
This is a very much oversimplified pedagogic model, but the goal is to show that when you propose a mechanistic pathway of a reaction, you must search
for the one with the lowest activation energy, because that is how molecules "decide" if they will react or not. If they have enough kinetic energy to
reach any possible transition state they will roll over the to the other side of the thermodynamic hill and give the respective product.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Sandmeyer
National Hazard
Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
Ok, since Arrhenius have not posted any problems for a while I'll post a problem. Provide mechanism for transformation 1 to
2 in DMF. I'll give a solution proposal on request.
EDIT: It is hard to see from the picture but it should read: "1.) Br2 2.) Base"

Gool luck!
[Edited on 24-12-2010 by Sandmeyer]
|
|
|
cheeseandbaloney
Harmless
Posts: 21
Registered: 4-4-2008
Member Is Offline
Mood: No Mood
|
|
yo sandmeyer! does it involve any tautomerization out of curiosity?
|
|
|
| Pages:
1
2
3
4
5 |
|