DrDevice
Hazard to Self

Posts: 74
Registered: 19-3-2012
Member Is Offline
Mood: Incompatible with carbon based lifeforms
|
|
Reduction and deacylation of ethyl acetylferulate with NiCl2/Zn
I've been performing experiments with derivatives of ferulic acid, an easily available cinnamic acid. An example with esterification and acylation is
described in
http://www.sciencemadness.org/talk/viewthread.php?tid=154722
I next wanted to reduce the double bond in the ethyl acetylferulate produced in that experiment using NiCl2/Zn. A similar reduction of ethyl cinnamate
is described in "Studies of Reduction of Various Organic Compounds with the Nickel (II) Chloride-Zinc system", (Nose, Kudo, 1990).
Typical methods in the literature for cinnamic acid alkene reductions use Pd and hydrogen, but that is not readily available to me. NiCl2/Zn is also
much cheaper :-)
I think I achieved the reduction, but I also think the acyl group has been eliminated, ie:
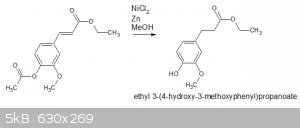
A question - should I have expected the acyl group to be removed? Any guidance on the mechanism would be much appreciated, and/or methodology that
would avoid the deacylation.
Procedure:
4.21 g (15.9mmol) ethyl acetylferulate is dissolved in 75ml MeOH, 7.7g (31.9mmol) of NiCl2.6H20 is added and stirred rapidly.
8.3g (127mmol) of Zn dust is added in portions. A small amount of hydrogen gas is evolved. The solution goes from green to grey/black fairly rapidly.
The mixture is then refluxed for 2 hours until all source material is gone (TLC, 1:3 EtOAc:hexane). One hour is probably sufficient.
The MeOH is then evaporated, leaving a viscous, slightly yellow liquid. This is re-dissolved in DCM, and washed with water and brine. An emulsion
formed that took some time to settle, but the brine eliminated most of it. EtOAc may be a better alternative - I will try that next time.
The organic phase is dried with MgSO4, and DCM evaporated, leaving 2.4g of a colourless viscous liquid. TLC showed a single spot, Rf = 0.53
vs 0.58 for the source material.
Vapour forms when the product is heated to the region of 125 - 130C - I haven't sufficient to get an accurate thermometer reading - compared to a
literature value for BP of 128C. The literature description is "a thick clear liquid". Assuming my identification of the product is correct, this is
10.7mmol, a yield of 67%.
|
|
|
clearly_not_atara
International Hazard
Posts: 2692
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
The reaction contains water and protons, which are sufficient to attack the ester. This side-reaction is difficult to avoid because the dissolution of
ZnCl2 byproduct releases protons from the water of crystallization which accompanies NiCl2:
NiCl2*6H2O + Zn >> ZnCl2(OH)2(2-) + 2 H+ + Ni + 4 H2O
To avoid this you would need to use an anhydrous form of NiCl2, possibly as the methanolate, and possibly an additional activating agent for the zinc
(although NiCl2 alone might be enough). There are some papers regarding nickel halide alcoholates:
https://onlinelibrary.wiley.com/doi/abs/10.1002/978047013244...
"One-inch-thick layers of hydrated nickel chloride are arranged in open Pyrex ovenware dishes which are then stored for 17 hours at 80" in a
circulating air oven, continuously purged with dry air. The resultant ycllow hygroscopic nickcl(I1) chloride dihydrate is pulverized in a Waring
Blendor and then transferred promptly to securely capped jars. The water content (Karl Fischer titration) varies from 1.88 to 2.2 hydrate."
"A three-necked, 3-1. flask fitted with a reflux condenser and mechanical stirrer is charged with 327 g. (2.0 moles) of nickel(I1) chloride 1.88
hydrate, 550 ml. of absolute ethanol, and 558 g. (4.14moles) of triethyl orthoformate.0 Thecream-colored slurry is stirred at the reflux under an
atmosphere of nitrogen for 2 hours, after which time the water content of a 1-ml. sample of the now homogeneous reaction mixture is checked with a
Karl Fischer titration. "
"* The Karl Fischer analysis should indicate that the solution is completely water-free. If water is present the mixture should be refluxed for an
additional hour. Another Karl Fischer analysis should then be performed, and if water continues to persist, an additional 0.1-0.2 mole of triethyl
orthoformate should be added and the refluxing continued
for an additional hour. Upon concentration of the solution to a volume of 1l., and then cooling it to 0" under nitrogen to exclude moisture,
pastel-green hygroscopic needles of the tetrakis-(ethanol) complex (140 g., 22%t) form. These are collectedand carefully dried under nitrogen on a
Schlenk frit at 23O. Anal. Calcd. for CsHZ4C1O4Ni: C, 30.61; H, 7.71; C1, 22.59;0, 20.39; Ni, 18.70. Found: C, 27.71; €1, 6.95; C1, 22.97;
0, 20.82; Ni, 19.09."
I assume that you could easily swap ethanol and triethyl orthoformate for methanol and trimethyl orthoformate or orthoacetate.
Trimethyl orthoformate is prepared by the reaction of chloroform and potassium methoxide.
Potassium methoxide may be prepared (in low yield) by dissolution of K2CO3 in anhydrous methanol. Surprisingly, the yield is around 50%, due to the
incredibly low solubility of KHCO3 in methanol, with higher yields obtained at low temperatures.
http://www.sciencemadness.org/talk/files.php?pid=492161&...
I suspect that the preparation of trimethyl orthoformate could be carried out in situ in the (filtered) methoxide rxn mixture, as the only
contaminant is (unreactive) K2CO3.
Trimethyl orthoacetate is prepared by the reaction of methanol and acetonitrile with hydrogen chloride. This reaction is more difficult to perform
(gaseous HCl) and is only preferable if you can buy acetonitrile, I think.
Overall though the use of NiCl2 alcoholates sounds rather difficult. One advantage would be -- if it does work -- it should reduce
nitroalkenes to aliphatic nitro compounds! Acidity causes the Nef reaction when NiCl2/Zn is used on these substrates.
[Edited on 24-1-2020 by clearly_not_atara]
[Edited on 04-20-1969 by clearly_not_atara]
|
|
|
DrDevice
Hazard to Self
Posts: 74
Registered: 19-3-2012
Member Is Offline
Mood: Incompatible with carbon based lifeforms
|
|
OK, thanks for the explanation.
I followed up with a bit more digging in the literature (yes, should of done that first...) to look at other deacylation approaches and it looks like
its pretty easy to remove with multiple techniques.
And I've done some follow up on the product to see if I can verify that it is the expected product, by taking the ethyl ester back to the carboxylic
acid and verifying the melting point. I'll post the experiment separately, but the melting points agree so I'm confident the point of the experiment
ie reduction of the double bond with NiCl2 / Zn worked.
|
|
|
|








