ChemichaelRXN
Hazard to Others

Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
TFDD 2,3,7,8-Tetrafluorodibenzodioxin
Hey,
Dont ever do this and I am not liable for your actions...I wont ever do this either, but I found the science interesting. I have no idea how toxic
this compound would be, being the fluoro analog. Nothing better than chloracne complexion lol
How toxic do you think it is?
One-pot reaction:
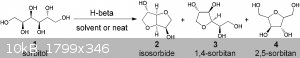
In a glass pressure vessel with teflon cap, heat a mixture of 4,5-difluorobenzene-1,2-diol with H-beta-zeolite. I dont exactly know how much, but it
should work as H-beta-zeolite is used for joining hydroxy groups and cyclizing.
I made bucky balls with H-beta-zeolite before, but never this pollution (dioxin).
I found it interesting though and thought Id share.
This is not a chemical weapon in Canada, I checked, so I shouldnt run into any issues talking about this, if that is even a possibility.
I would be interested if someone does this research one day, the bromo, iodo, fluoro analogs of dioxin, instead of TCDD and documents toxicity, but
who wants to handle this anyway.
Anyway, I will be posting a chemistry video of 3-methoxy-beta-nitrostyrene next and the reduction using gallium amalgam. I am an innovator, so it
carries along to chemistry too. I couldnt find any info on the reaction I did. I used gallium chloride instead of mercury chloride, which is
toxic...again, I dont handle anything too toxic.
See the attached images for an example of how H-beta-zeolite is used. You can see it attaches diols and I am sure some dioxin would be formed with
this method. (Just hypothetical and interesting.)
[Edited on 26-9-2020 by Mr.StinkyGas]
|
|
|
Pumukli
National Hazard
Posts: 686
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
What is the proper way of depressurizing such a vessel in the end? Is it only for reactions which don't end up with elevated pressures?
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
You might be able to chill it with dry ice and use appropriate solvent to wash it out under some sort of fume hood. I wouldn’t ever make it. You can
make a solution with what is added and remove a bit for testing too. I am guessing toluene might work and it wont spread it around the air as much
because of the boiling point, but I am guessing you would know all of this already. Different halogen substitutions would be interesting!
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
The nitrostyrene reduction with Al/Ga did work for your? Really?
So far, everything I've seen and read regarding gallium amalgam in comparison with plain mercury amalgam, did point into the direction that it just
doesn't work the same, or nearly as good or at all than Al/Hg.
You make this sound like you did it already, did you really?
How have you prepared the gallium chloride if so?
How have you amalgamated the aluminium, in what solvent?
What kind of aluminium have you used, grains I suppose?
Please describe the reaction, I'm more interested in that than seeing a video of it.
And please more details on that reaction, that is the first time I ever heard and read of this being possible in reality besides some pipe dreams!
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
No problem...I did the reaction, the aluminum broke down maybe a bit slower and you needed more gallium chloride in relation to mercury chloride, but
the reaction carried on with some heat applied and after separating the isopropanol layer when you neutralize it, I have a HCl solution in a beaker I
still have to purify and rotovap. I will let you know the yield. I started with 8grams of the 3-methoxy-beta-nitrostyrene, 50 ml Glacial acetic acid,
80ml isopropanol, 25ml distilled water, a solution with about 3grams of Gallium Chloride and 10g of cut squares of aluminum.
I neutralized with 200ml of water and 78g KOH.
You get a dark isopropanol layer to purify. I notice chloroform is good to remove the stuff you dont need, leaving the HCl solution to dry and
recrystallize. I will probably use it for the next step as-is though.
Again, I cant say anything until I dry the solution, so I am still in the middle of things.
Pictures below: 1) Henry reaction of 3-methoxybenzaldehyde to the 3-methoxy-beta-nitrostyrene
2) the gallium amalgamation
Making the gallium chloride is such a time consuming process of boiling 6g gallium with HCl that you have to continue adding (definitely need a fume
hood). There must be a better way. I still have some solution if I want to try it again.
Just make sure to add all the aluminum with stirring all at once because it wont react nicely without doing that.
 
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
Interesting!
Gallium chloride is said to react violent with water, but it still worked as aqueous solution for you?
Maybe that only counts for the anhydrous salt...
And you did the amalgamation in water?
Ok that is really interesting, I am eager to hear how it turned out.
That could potentially have an interesting impact on a certain niche of hobby chemists, if it has worked out fine.
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
I think it worked, but I will get back to you on this. Yes, it was with the aqueous solution of gallium chloride. I couldnt crystallize it because it
is hygroscopic and soluble in everything lol
I like the method too so far... it is much less toxic.
https://link.springer.com/article/10.1134/S0020168512020069?...
[Edited on 2-10-2020 by ChemichaelRXN]
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
Well, I have prepared gallium amalgam too years ago, by simply dissolving aluminium foil pieces into gallium, but that stuff was hardly as reactive as
I am used to from the normal Al/Hg.
It is interesting that it seems to be different when activated by a gallium chloride solution, that it even worked makes me wonder.
Ok I will now just wait eagerly to hear any updates on how it worked out in the end!
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
Ok, no problem, I will let you/everyone know.
(Yea, I am guessing the gallium isnt as volatile like mercury where the mercury from the mercury chloride doesnt jump from aluminum piece to aluminum
piece, so that is why you need more gallium chloride to cover all the aluminum from the start. You can see the discolouration as if it is mercury on
the aluminum from the reaction of the gallium depositing and a nice steady hydrogen production, aided with heat.)
[Edited on 2-10-2020 by ChemichaelRXN]
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
After looking a bit around, I think that sadly your reaction probably hasn't worked as intended.
Yes, the aluminium has reacted, but not in the right way.
Because the equation for gallium chloride in water is this: GaCl3 + 2H2O → Ga(OH)2Cl + 2HCl.
I suppose what you have seen was simply the produced HCl eating up the aluminium.
I don't know how fast that reaction is, but it is stated everywhere that GaCl3 reacts violently with water, so I assume that it is not very slow.
But I would be happy if I'm proven wrong and you can indeed confirm a successful reduction!
I guess it would be safer, to amalgamate the aluminium with the GaCl3, and when it has been amalgamated successfully, to simply draw the solution off
of it, wash the activated Al once with water or alcohol, and then add the alcoholic solution off the substrate with some acetic acid and to run the
reduction under continunal readjusting the temperature, etc.
This way one could at least avoid the potential iissues that could result from the reaction of GaCl3 with water, as HCl really does interfer
negatively with that reduction.
I mean, for Al/Hg we only keep the Hg salt in solution as it doesn't hurt to have it in there, but it could also be remvoed easily as some tend to
activate the Al and then pour the solution off when its done.
This should work for Al/Ga too.
I'm really excited to hear results! 
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
I was under the impression that as if Zn/HCl the Aluminum producing hydrogen like it did will reduce the 3-methoxy-beta-nitrostyrene...you are
probably right and maybe it doesnt work so well for phenyl-2-nitropropenes with leaving the GaCl3 in solution, but I wont be producing any
amphetamines as they are all scheduled in Canada. Pouring off the gallium solution after activation will probably work because you can tell some
discolouration of the aluminum when it starts reacting as if soaked with gallium and producing hydrogen with the water added in the reaction.
Anyway, I will let you know!
I was actually thinking of running just HCl and Aluminum for the nitrostyrenes because you might be able to separate it easier, but not sure. The idea
after neutralization would be to easily separate out the isopropanol added (with the acetic acid conditions added as well if you want to test out for
phenyl-2-nitropropenes, but maybe I am wrong)
[Edited on 2-10-2020 by ChemichaelRXN]
You are the same perception looking out, from the same elements around the universe.
You are everything to be anything to begin with.
Https://you-are.space
Https://syntharise.com
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
The issue would be that simply Al dissolving won't result in nascent hydrogen which is why the amalgamated Al works for reduction but not simply
dissolving Al in acid or base.
Well, we will see how it turned out!
And oh no, I don't want to reduce phenyl-2-nitropropene with it
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
Lol Yea I definitely cant though.
It is still a nitro and a double bond for both beta-nitrostyrene and phenyl-2-nitropropene in the case of Zn/HCl....maybe less acidic conditions are
needed
You are the same perception looking out, from the same elements around the universe.
You are everything to be anything to begin with.
Https://you-are.space
Https://syntharise.com
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
In case of Zn/HCl, they reduce different.
The one goes to the amine, the other to the oxime.
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
In acidic conditions with Zn/HCl you get the oxime, basic conditions, like; triethylamine (if i can recall correctly) you get the amine, but a beta
hydroxy for the compound; phenylpropanolamine. Just in the “middle”, with a weak acid (acetic acid) and aluminum hydroxide forming to the acetate,
you get the amine only (Al/Hg and hopefully Al/Ga too!) It is interesting chemistry. I wish I knew a bit more in depth as to why.
I will be purifying the solution soon, so by next weekend when I am not so busy, I will let you know the yield. I notice with a 2,3,4,5-tetramethoxy
as opposed to a 3,4,5-trimethoxy the affinity increases. I am not exactly going to be consuming this research compound (3-methoxyphenethylamine) i am
making, but it was a fun experience with the gallium chloride anyway. I would be curious to know what the affinity is like though. The 3-methoxy group
is known to have affinity for serotonin...i might have mentioned this before. I am interested in the 3-MeO-NBOMe HCl...
[Edited on 2-10-2020 by ChemichaelRXN]
You are the same perception looking out, from the same elements around the universe.
You are everything to be anything to begin with.
Https://you-are.space
Https://syntharise.com
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
Its the double bond besides the alpha-alkyl group which causes that you end up with an oxime always when you attempt to reduce a phenylnitropropen.
You cannot get to the amine with them, as these reducing agents do not reduce oximes.
Unlike nitrostyrenes which do end up with the amine.
If it is just the nitro group without a double bond besides and an alpha-alkyl chain, then you get the amine in all of the mentioned conditions.
I don't understand what you mean with triethylamine and reduction, this is not about the nitro aldol reaction but about the reduction of their
products?
And clearly the nitroalcohol you apparently do mean has no double bond, this is why it can be reduced to the amine, despite its alpha-alkyl chain.
That has nothing to do with basic conditions, which are by the way the formation of the nitroalcohol.
Not its reduction as that is done in acidic conditions AND has also been done in basic conditions like with nickel boride 
Think of phenylnitropropanes, they get reduced with Zn/HCl to the amine too and thats acidic.
But, with NaBH4/NiCl2 or NaBH4/CuCl2, you get the amine from them too, but in basic conditions again.
But not the nitropropenes which give oximes, that is because of the double bond and the a-alkyl group solely.
Also, I know a basic Al/Hg in presence of ammonia well, that reduces oximes to amines.
But the standard method is an acidic Al/Hg in presence of acetic acid, oximes to amines.
I hope you do understand your initial misconception?
Sadly next weekend only?
But you said you already have the basic IPA extract? Wash that extract with saturated K2CO3 solution, this removes most of the water from it, maybe do
it twice.
Then you can just form the oxalate which precipitates quick, clean and simple, thats less than an hour work until you have it isolated and dry!
Suction filter the precipitated oxalate, wash with acetone at the pump, then suck dry, and you know the yield.
All in all one hour.
And then we know if it has worked, and if so, also the yield.
Come on, thats not too much to ask?
I really want to know if it has worked, not how much the yield is, at this point it is only if it even worked as you intended. Or if you just
dissolved aluminium with in-situ produced HCl, which is what I am now slowly start to get convinced was the more likely outcome.
In that case, it could be an explaination for your discoloured IPA layer.
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
Yes, I understand. I havent studied those reactions for quiet a while. There was a reaction I could slightly recall where you you use
triethylamine...i was incorrect about it though (it was for the 1-phenyl-2-nitroalcohol for Phenylpropanolamine) nevermind...sorry about that!
What is the method for NaBH4/CuCl2 by any chance? Maybe PM me or post it here, whatever seems appropriate.
Anyway;
I have a solution of isopropanol and a dilute HCl solution. It precipitated very slight tar on the walls of the flask, lightening the solution up. I
have to simply rotovap for a crude solution or wash with chloroform first and then with the aqueous solution, I rotovap. It shouldnt take me long. I
will do this tomorrow and let you know.
You are the same perception looking out, from the same elements around the universe.
You are everything to be anything to begin with.
Https://you-are.space
Https://syntharise.com
|
|
|
monolithic
Hazard to Others
Posts: 435
Registered: 5-3-2018
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by ChemichaelRXN  | | I have a solution of isopropanol and a dilute HCl solution. It precipitated very slight tar on the walls of the flask, lightening the solution up. I
have to simply rotovap for a crude solution or wash with chloroform first and then with the aqueous solution, I rotovap. It shouldnt take me long. I
will do this tomorrow and let you know. |
Any updates on this? Al/Ga reaction is interesting if it works.
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
Sorry to keep you waiting. I will have to go with what I previously said and will let you know on the weekend.
You are the same perception looking out, from the same elements around the universe.
You are everything to be anything to begin with.
Https://you-are.space
Https://syntharise.com
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
Ok, well it isnt that great. Something to work on or improve and I will try again one day or use another method because I want
3-methoxyphenethylamine. The yield is not lower than 30% in this attempt and I did get a bit of tar precipitating when the solution was kept in the
fridge, which I separated before rotovaping.
So the gallium chloride experiment didnt go that great. Sorry about that. I did get some 3-methoxyphenethylamine HCl, but not enough! Back to work!
[Edited on 8-10-2020 by ChemichaelRXN]
You are the same perception looking out, from the same elements around the universe.
You are everything to be anything to begin with.
Https://you-are.space
Https://syntharise.com
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
Ok, interesting.
But why had it the amine odour after the neutralisation?
Oh and please isolate the amine from the product you obtain now, as I am sure some part is composed of the oxime and probably unreacted substrate too,
due to your workup.
I recommend to wash the aqueous solution of the supposed HCl salt with 2-3x portions of DCM first(I am sure some of the precipitate will go into
solution, i.e. that would indicate it is some side product).
The amine HCl will not precipitate from water, that is something you can definitely believe me, not even from 3-4ml, it will only appear as a solid
when the water is completely evaporated.
And when the solution is clean and without precipitate from the washings, you can freebase it, and take the base up into DCM, ether, toluol or what
you got, but not IPA.
And then I would, as I already recommended, form the oxalate salt via precipitation from the solvent(add an acetonic or isopropanolic solution of
oxalic acid to the freebase in solvent would be easiest).
The oxalate will be clean and white and form fast without problems.
At the same time, the precipitation from solvent will ensure that anything unwanted stays in solution.
Filter and dry the oxalate off, and then you can access the yield more realistic, and oh, you should also assume that you have the hemifumarate, so 2x
amine molecules on 1x oxalic acid.
Your isolation method is not good, I mean extraction with IPA is alright, thats true, but the way you tried to get your amine from the IPA is really
nothing to recommend.
The best way is to precipitate the amine from the IPA as salt, sulfuric acid is used sometimes for this for example.
But again, I strongly recommend oxalic acid instead, it is a wonderful tool for the work with amines and often underrated for its usefulness, but it
deserves a lot more attention really.
You can without any problems use the oxalic acid in slight excess and this will not result in any issues, since oxalic acid is soluble in IPA and
doesn't requires to be exactly titrated like sulfuric acid does.
And, as said, oxalates have the huge advantage to form always readily, almost instantly, and they have that nearly unchallenged property to come out
very clean, no matter how clean the extract solution was.
In the future, try my method instead, it is the superior method to isolate amines from IPA extracts, no matter if coming from an Al/Hg or NaBH4/CuCl2
which both use IPA as reaction solvent, and so it is simply convenient to use it as extraction solvent as well.
And in that case, oxalic acid is the perfect way to get the amine out of the IPA without having to deal with all the other crap that is dissolved in
the alcohol too, just acidify and cool it down, the oxalate will readily but slowly settle on the bottom, as a fine powder usually.
The oxalate can then be used to freebase the amine out again, and either (steam) distill or solvent extract the freebase again, and you will notice a
greatly improved purity at that point, not to mention on the finally produced salt of the amine.
However, the oxalate is already fine as final product, in case the amine is not intended for human consumption.
|
|
|
ChemichaelRXN
Hazard to Others
Posts: 103
Registered: 7-10-2010
Member Is Offline
Mood: Universal Eye
|
|
Thanks for the information. It was a rough precipitate, that’s correct. I will be adding chloroform to it to remove any residues and then dissolving
product in water, adding NaOH and then a chloroform extract which I can get the product from. DCM would possibly dissolve the product if I can recall
correctly as the HCl salt. It was my first attempt, but I will consider precipitating it as the sulphate right from IPA or just move on to NaBH4 and
CuCl2. I guess I can also use fumaric acid right from the IPA too. (I dont have oxalic acid, just fumaric) There is definitely some product their, but
it needs improvement and I need more time because I have a busy schedule.
I will make a separate thread when I retry and if I get a better yield.
[Edited on 8-10-2020 by ChemichaelRXN]
You are the same perception looking out, from the same elements around the universe.
You are everything to be anything to begin with.
Https://you-are.space
Https://syntharise.com
|
|
|








