SuperOxide
Hazard to Others

Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
First mononitrotoluene synthesis - Having some problems (I think?)
I'm doing my first nitrotoluene synthesis, just a quick summary of how it's gone so far:
- The mix was kept at below 10 °C (I know most say 15 °C, so figured this would be fine).
- The nitration mix was added drop by drop, over the span of a few hours.
- The reaction mix went from clear, to yellow, to red, to very dark red, then to a mostly orange (with some red at the very top). From what I
understand, it should go back to being yellow, signaling the end of the reaction.
- I let it stir at 10 °C for almost 2 hours past when all the nitration mix was added (so this is like 4 hours into it), and it still hasn't turned
yellow. Just saying orange.
- I figured "Well maybe the 'end of the reaction' is when I let it slowly warm up to room temp - So I let it slowly warm up, but nothing changed.
- When it hit 19 °C and didn't change at all, I started to cool it back down again, it's now at 13 °C (I panicked, worried that some di-nitrated
product may form).
- I think one of the problems is my damn stir bar(s). I've tried a few, but they all seem to dance around if I go too fast (usually I can find a way
to fix that, but this time it's more stubborn than usual).
Here are a few photos of the current state: https://imgur.com/a/WkXBWkq
(I usually download those then scale them down so I can attach them, since they're far too large to insert, but I'm trying to pay attention to the
reaction atm, sorry).
Questions:
1) Is this because the stirring isn't at MAX? It's more at like the lower side of medium speed... Not enough to get the two layers to mix 100% (hence
the red layer on top of the orange still).
2) Did I fuck up by letting it warm up to 19 °C (for ±20 minutes or so)? Will that result in some di-nitrated toluene that may blow me to
smithereens?
3) Should I keep on stirring this until it turns yellow?
P.S. sorry for the haphazard style, just busy babysitting this thing.
[Edited on 28-2-2021 by SuperOxide]
|
|
|
SuperOxide
Hazard to Others
Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
Update 1: It's now turning a lighter orange (still orange though.. but less red, I guess). So i suppose my shitty stirring just means
I needed to give it time to react...
My main 3 questions now are...
1) Is this mainly because of my less-than-stellar stirring performance? That's my bet..
B) Could keeping the reaction mix <10 °C (even closer to 5 °C for a decent portion of it) have caused it to take much longer as well?
III) Is my house going to blow up (sarcasm) because I let it warm up to 19 °C for 20 minutes? Or is it just going to effect the purity?
Thanks guys.
Update 2: Going on like 6 or 7 hours now, just letting it get to room temp. I think I would be ok calling it "yellow" now. It just
took for-fucking-ever.
And I found out why my stirbar wasn't working out so well. The tubberware I was using for the ice bath, it has this little rim thingy on the bottom
that's literally like 2 or 3mm high... Apparently that's too damn high for my hotplate/stir bar. Good to know >_<
[Edited on 28-2-2021 by SuperOxide]
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
I kept nitration at 0-5 C and although that I obtained some dinitroderivates. I even tried vacuum distill them, first mononitro distilled out and
finally dinitro but that was slow and I stopped it soon (for dinitro my oil bath reached temp 180-190 C, and I used 2 stage vacuum pump with an old
oil so the vacuum was probably not so good as the theoretical value 0,3 Pa claimed in the documentation).
Washing with 1% NaOH removes oxydation sideproducts and improves color.
Dinitro if any is only in tiny amount, very diluted. Even if pure dinitro it does require initialization to undergo detonation.
The best laboratory preparation I have ever read is in a link in this post:
https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
[Edited on 28-2-2021 by Fery]
|
|
|
SuperOxide
Hazard to Others
Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
Ok, so it's all finished.
Basically it took forever because the container I used for the ice bath really hindered the stirring... So it went on for like 8 hours (more,
actually), and eventually turned a yellow. Not the same color I would like it to have been (as seen in other photos and videos), but I settled for it,
hopefully it doesn't hurt the yield too much (haven't measured it yet).
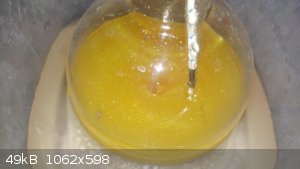
I put all three isomers into a small jar and put it in the freezer over night to see if it would freeze. I know it's dry, so I figured if there were
too many impurities then perhaps it wouldn't freeze, but it froze just fine. So I will separate them today.

Well, that was one hell of a learning experience, lol. Glad I at least got something out of it though. I thought I botched the entire synth up.
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
Well done!
Back when I did this reaction (w/ 102g toluene, 217g conc. H2SO4, 153g 60% HNO3), I gave it four hours starting at 1°C, for the remainder hovering
between 4-8°C, and ended up with a dark red top layer. Most of the remaining toluene had to be distilled off azeotropically, and the last bit steam
distilled along with a small portion of the product.
After that mess, and after freezer separation, 39% crude yield of o-NT, 21% of p-NT.
I suggest getting some Bel-Art brand stir bars, they're easily the best I've used, I believe Corning ships their hotplates with those.
Do post the amounts of each reagent used!
|
|
|
SuperOxide
Hazard to Others
Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
Quote: Originally posted by Benignium  | | I suggest getting some Bel-Art brand stir bars, they're easily the best I've used, I believe Corning ships their hotplates with those.
|
I'm 100% sure that it was just the bottom of that container I was using for the ice bath. It kept the flask just high enough off the hotplate that the
magnetic connection was attenuated too much. I think I may have ran into this with my anthranilic acid synthesis (using the same setup), but I just
assumed it was because there was a lot of precipitate on the bottom throwing the stirbar off.
Sure thing (this was also the first chemical experiment/reaction where I actually wrote the reaction/results/notes/suggestions in my "lab journal"
 ) )
I used:
75mL conc. nitric acid
90mL conc. sulfuric acid
78ml purified toluene
Still haven't separated the isomers yet, but I may post the yields when that's done. I do plan on doing this again next weekend, I may up the scale
slightly, but I can't too much, as it's limited by the glassware I have.
On a side note, when I did the sodium bicarbonate wash in the sep funnel, it formed what I thought was the most pretty emulsion I've had to deal with
yet. I had to take a picture.

However, it turns out it wasn't at all an emulsion (which I waited like 3 hours to let it separate before I realized this, just more time wasted), but
a sign that I need to stop using tap water to clean the glassware. I think that's just the two phases beading up on the inside of the glass. So bad
sign, but really pretty photo, still.
Edit: I suppose I shouldn't have said "just more time wasted", as I learned something. As long as I learn something new (no matter
how small), I don't usually consider it a waste of time.
[Edited on 1-3-2021 by SuperOxide]
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
Great time to start keeping complete notes! It was really hard for me personally to start writing stuff down, but as it turns out, being able to
recall every step in detail is strangely addicting.
Beautiful colors indeed! Two things I'd suggest: Get a cheap reverse osmosis filter that can run on the pressure of municipal water, 2 bar or so.
Great for producing clean water to rinse with, as well as for reactions. I've had one for many years now and the membrane is still good, though if
there's chlorine in your water you may have to swap it for a new one every couple of years. Also, a base bath is often the best solution for etching
glassware clean of residue and making it uniformly hydrophobic.
There is indeed immense value in things not going according to plan and outright failure. Good on you for appreciating it. 
|
|
|
SuperOxide
Hazard to Others
Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
| Quote: Originally posted by Benignium | | Great time to start keeping complete notes! It was really hard for me personally to start writing stuff down, but as it turns out, being able to
recall every step in detail is strangely addicting. |
I have kept some notes before, usually in a
text editor on my Macbook. But they weren't complete details, it was more for when I was repeating an experiment/extraction, writing down the
quantities and results, then the yields (eg: I repeated the anthranilic acid synth quite a few times, kept some of those notes). But I just got a neat
notebook that I actually write in now (the synthesis planned, the procedure to follow, the quantities used, and then I notate the changes as I go
along with the times next to them). I can already see it will be very helpful!
| Quote: Originally posted by Benignium | | Beautiful colors indeed! Two things I'd suggest: Get a cheap reverse osmosis filter that can run on the pressure of municipal water, 2 bar or so.
Great for producing clean water to rinse with, as well as for reactions. I've had one for many years now and the membrane is still good, though if
there's chlorine in your water you may have to swap it for a new one every couple of years. Also, a base bath is often the best solution for etching
glassware clean of residue and making it uniformly hydrophobic. |
For sure. I plan on doing that sometime in the future. I suppose for now I will just do the last rinse with distilled water, and give the base bath a
shot (I rarely do base/acid baths for cleaning, but I need to raise my standards for cleaning glassware, obviously).
| Quote: Originally posted by Benignium | | There is indeed immense value in things not going according to plan and outright failure. Good on you for appreciating it. |
Absolutely. I've always maintained that
you learn more from being wrong than you do from being right. If you aren't making mistakes, then you aren't trying anything new (Elon Musk said that,
but Im sure he got it from someone else).
|
|
|
mr_bovinejony
Hazard to Others
Posts: 121
Registered: 20-4-2018
Member Is Offline
Mood: ASS
|
|
Does the quality of water really matter so much? I got sick of buying gallons of distilled water so I've been using tap water for reactions and for
cleaning and haven't noticed a difference in any of my recent projects
|
|
|
SuperOxide
Hazard to Others
Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
| Quote: Originally posted by mr_bovinejony | | Does the quality of water really matter so much? I got sick of buying gallons of distilled water so I've been using tap water for reactions and for
cleaning and haven't noticed a difference in any of my recent projects |
I suppose it really depends. There are some reactions where it absolutely does matter, like the hydrazine sulphate synthesis (I believe tap water has
a high amount of metal ions that don't all get chelated properly), I know there are other examples, but this is the only one that comes to mind. Also,
when you use nice clean water, you're removing a variable (impurities). If a reaction fails, you can be sure it wasn't due to impurities in the water
(plus that would be a crappy thing to have a reaction fail for, something so simple and easily preventable).
I use to sometimes use the bottled drinking water from Walmart, but then I noticed on the ingredients it actually has some calcium chloride and sodium
bicarbonate, which could also interfere with reactions (eg: titrating an acid to determine the purity could be hindered by the unknown amount of
sodium bicarb in the water).
In terms of cleaning glassware, I use tap water, but tap water will definitely not pass the water bead test - which is basically what seems to have
happened inside my sep funnel, the two different layers stuck to the glass in the incorrect phase because the liquid "beaded" (ie: clung to) the
impurities on the inside of the funnel, but when I drained the funnel the two layers came out separately.
So basically, it depends on the reaction and just how clean you need it to be.
[Edited on 2-3-2021 by SuperOxide]
|
|
|
SuperOxide
Hazard to Others
Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
For anyone wondering, I got the two isomers separated (kinda). The liquid (before separation) was 74.53g (67 mL), and after
separation, the ortho isomer weight is 54.31g, and the para isomer is ±17g.. so I somehow lost
over 3g during the freezing/vacuum filtration due to mechanical losses. It's a little difficult to avoid that since I'm not sure what I could use to
wash out the filter/beaker/graduation cylinder/etc with, so when I pour/scoop the contents out, some is still left (and I repeated the freezing point
separation at least 6 or 7 times). I suppose having a scale with questionable accuracy could also contribute to this.

You can see the para-nt is still evidently wet with ortho-nt because I had to do the vacuum filtration with a hand pump (my forearms
haven't had such a good workout since I was a teenager). I will definitely need to do a better job on the filtration somehow. However, I only really
care about the ortho isomer right now, so as long as I get as much of the para out of it as I can, I don't mind if the para is a little
contaminated with the ortho.
I plan on doing this synth again on a slightly larger scale (and with better chilling and stirring).
[Edited on 4-3-2021 by SuperOxide]
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
very nice!!!
recryst para from methanol
your liquid is very likely still a mixture of ortho and some amount of para
|
|
|
SuperOxide
Hazard to Others
Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
Thank you. It was a lot of work, but also a lot of fun. I def plan on doing this again. Likely
this weekend, on a slightly larger scale (not double or anything, maybe more like 125%).
I was just looking at the PubChem thinking maybe I should try this. I was going
to try it with chloroform, but maybe methanol would be a better choice.
Yeah, I have that suspicion as
well. I know that if I can apply a better vacuum/suction then it will filter off better before the para can dissolve into the ortho, perhaps I'll
jerry-rig the shopvac to it (I got that idea from an Extraction&Ire video, lol).
Other than distillation (since apparently it's nearly impossible to separate the two isomers that way), you have any other suggestions?
Also, after I do the recryst for the para using MeOH, would it be somewhat safe to just boil off the MeOH from the liquid, and mix that back into the
ortho liquid? Or do you think it would contain more impurities than just the ortho isomer?
Edit: I just saw a post in this thread suggesting using a centrifuge on the mixture while it's frozen. I have a [crappy] centrifuge, maybe that's worth trying? I could fill
the tubes, put them in the freezer, then run the centrifuge as it warms up. I don't see how the para would stay as a solid at the bottom though, since
it would have the ortho liquid right above it to dissolve into. What do you think?
[Edited on 4-3-2021 by SuperOxide]
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
Good stuff!
What I and many others have done when filtering out the p-NT, is rinsing it with alkanes in which it is insoluble or the solubility is negligible. I
recommend doing that, followed by dumping it out on a coffee filter with some layers of paper towel underneath to wick away most of the solvent, then
moving the whole thing to a well ventilated spot so the rest can evaporate. I haven't tried the MeOH recrystallization myself, but I would still
encourage you to follow up with that to improve purity.
When filtering, it helps a lot to also put the büchner or whatever you filter with in the freezer as well.
As for the vacuum source, I think you should have no problems with this particular filtration using the handheld diaphragm pump, but I would strongly
recommend into getting a high-powered water pump that can match the pressure and flow rate of municipal water. Get a $10 aspirator pump from here and some tubing with 16mm & 10mm ID and you've got a very respectable and durable vacuum pump that, depending on the temperature of water
used, can go to 50-75 mmHg. NurdRage has a nice video on the subject, but beware of pumps that small. I've tried two and a high cut-off pressure alone will not cut it, there has to be
considerable flow. Another thing I'd recommend is to try to get the vacuum take-off port welded onto the rest of the aspirator, since the particular
one I linked, at least, has pretty weak threads there and those will likely corrode and break off in a year or two.
Give the centrifuge a shot since you have one! It may or may not be worth the trouble but perhaps check to see if its operating temperature allows for
it to be chilled beforehand as well?
|
|
|
SuperOxide
Hazard to Others
Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
Benignium - nice to hear from you, as always!
| Quote: Originally posted by Benignium | | What I and many others have done when filtering out the p-NT, is rinsing it with alkanes in which it is insoluble or the solubility is negligible. I
recommend doing that, followed by dumping it out on a coffee filter with some layers of paper towel underneath to wick away most of the solvent, then
moving the whole thing to a well ventilated spot so the rest can evaporate. I haven't tried the MeOH recrystallization myself, but I would still
encourage you to follow up with that to improve purity. |
Yeah, I plan on doing that as well. I started
letting it dry out in a coffee filter with a fan over it, but it started to get a bit stinky, so I thought maybe I should wait til I have the house to
myself before I do that.
Oh.. When I do the MeOH recryst, I should be able to pour off the liquid that didn't crystalize and just evaporate the MeOH off, leaving the
"impurities" which are likely just mostly ortho product, right? I'm asking because the ortho product is what I'm going after, so if I can recover some
from the recryst step, then thats a bonus.
Great idea. The videos/threads I've read on the subject suggest taking the NT mixture out of the freezer when it's totally frozen, mixing it up, then
letting it cool down to just above -10 °C before putting it in the funnel. I thought it would be better to just put it all in the buchner funnel
asap, then apply vacuum to pull off the ortho product as it melts (I guess so it doesn't have a chance to dissolve some of the para product
into it). Seems to work ok.
| Quote: Originally posted by Benignium | | As for the vacuum source, I think you should have no problems with this particular filtration using the handheld diaphragm pump, but I would strongly
recommend into getting a high-powered water pump that can match the pressure and flow rate of municipal water. Get a $10 aspirator pump from here and some tubing with 16mm & 10mm ID and you've got a very respectable and durable vacuum pump that, depending on the temperature of water
used, can go to 50-75 mmHg. NurdRage has a nice video on the subject, but beware of pumps that small. I've tried two and a high cut-off pressure alone will not cut it, there has to be
considerable flow. Another thing I'd recommend is to try to get the vacuum take-off port welded onto the rest of the aspirator, since the particular
one I linked, at least, has pretty weak threads there and those will likely corrode and break off in a year or two. |
Yeah, a vacuum aspirator is definitely on my "buy asap" list (along with a better water pump, 1L 2neck flask, and a 24/40 to 14/20
adapter, lol). The water aspirator is one of those things that I know I need, but whenever I get the chance to spend a little $ extra on something, I
get chemicals instead, haha.
| Quote: Originally posted by Benignium | | Give the centrifuge a shot since you have one! It may or may not be worth the trouble but perhaps check to see if its operating temperature allows for
it to be chilled beforehand as well? |
I just did. The centrifuge I have lets you take out the little tubes
that hold the actual test tubes. So I wrapped the test tube in a paper towel, inserted it into the centrifuge tubes, wrapped them up and put them all
in the freezer over night. And I think I must be doing well with the separation because only 2 of the 4 I put in the freezer had any solid in
them at all, and it was very little. And after I put them in the centrifuge and then vacuum filtered it all, it was hardly anything left in the
filter. So either the centrifuge trick didn't do well or there wasn't much para product for it to freeze.
I plan on nitrating maybe 100mL of toluene this weekend, so I will have plenty momo-nitrotoluene to test it out again later in the week. I will update
the thread with results.
[Edited on 5-3-2021 by SuperOxide]
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
I actually don't think you'd recover much o-NT from evaporating the methanol, if at all. In theory, there is going to be some inside the crystal
structure of your p-NT, as well as molecules that didn't get rinsed off the surface after filtration, but unless you were to reuse the methanol for
consecutive recrystallizations, I doubt the exercise would be worthwhile.
Looking for a water pump for condenser water? I suggest scavenging for old 9-12V DC power supplies and getting a couple of these:
https://www.ebay.com/itm/DC12V-3m-240L-H-Ultra-Quiet-Brushle...
For the price they're awesome. They're durable and can withstand quite a bit of resistance; I've used one to supply two condensers at the same time,
and I have one fitted with a chainsaw fuel filter, to prevent the outdoors from clogging it.
|
|
|
SuperOxide
Hazard to Others
Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
So I just re-did the synthesis keeping everything the same except for the scale (just 25% larger scale) and the stirring. And it was definitely the
stirring that did me in last time. I was able to put the stir speed on max, and got the entire thing done in less than half the time
| Quote: Originally posted by Benignium | | I actually don't think you'd recover much o-NT from evaporating the methanol, if at all. In theory, there is going to be some inside the crystal
structure of your p-NT, as well as molecules that didn't get rinsed off the surface after filtration, but unless you were to reuse the methanol for
consecutive recrystallizations, I doubt the exercise would be worthwhile. |
Ok, good to know. I haven't done
the recryst yet, but I did put the "wet" pNT on a towel for a few hours, and now they're nice crystals, with just a hint of yellow (which is normal I
believe).
Heres a quick photo:

The dried crystals are on the left, the wet ones on the right are from todays synthesis, I need to do the freeze/filter cycle a few more times before
I dry them (few other photos).
I'm not sure what I'll use the pNT for yet (maybe p-Nitrobenzoic acid? not sure), but I will recrystallize it all after I've combined it with
the pNT from this run.
| Quote: Originally posted by Benignium | Looking for a water pump for condenser water? I suggest scavenging for old 9-12V DC power supplies and getting a couple of these:
https://www.ebay.com/itm/DC12V-3m-240L-H-Ultra-Quiet-Brushle...
For the price they're awesome. They're durable and can withstand quite a bit of resistance; I've used one to supply two condensers at the same time,
and I have one fitted with a chainsaw fuel filter, to prevent the outdoors from clogging it. |
That's what I
am going to get. I (stupidly) ordered this water pump from Amazon without doing much research on it, and it's actually not submersible. It also has a ridiculous amount of power for
such a little pump, if I use the hoses I typically use for water circulation it just collapses the tube on the input side of the pump (3LPM, 100PSI...
It's meant for RV's and boats, wtf was I thinking? lol).
So I'm returning that one now, then getting a nice submersible pump like I should have gotten in the first place.
[Edited on 7-3-2021 by SuperOxide]
|
|
|
|









