smaerd
International Hazard

Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
n-amylbenzene Preparation
Disclaimer I have never taken an organic chemistry class, try not to be too harsh.
So it is my intention to investigate the products(isomers) from the original Friedel Crafts reaction, amylchloride, benzene, and AlCl3(iirc). The goal
is to use old-school analytical techniques. In order to do so, I will have to develop standards, to compare products against.
Org. syn. has a procedure for n-amylbenzene, however it uses a pretty rare chemical, that requires a rare chemical or three to make. So it is sadly
out of the question. After consulting with a couple chemical supplies, the lowest price I can find for n-amylbenzene was about 50$ for 25 grams of 98%
purity(yellow liquid? references say it should be colorless). I'm simply too cheap for this  . Why spend 50$ for an excess of a chemical when I can spend 30$, and have a couple useful left-over chemicals and the
experience of preparing it? . Why spend 50$ for an excess of a chemical when I can spend 30$, and have a couple useful left-over chemicals and the
experience of preparing it?
A few papers have helped to light the way, however I can't seem to find this one's abstract on any of the popular journals: Morton and Fallwell, Jr.,
J. Am. Chem. Soc. 60, 1429 (1938).
Anyways... I'm babbling again... The scheme I've concocted from relatively easy to acquire materials is attached and discussed below. The idea of
which came entirely from:
| Quote: | n-Amylbenzene has been prepared by the action of sodium on a mixture of benzyl and butyl bromides;1 by the reaction between benzyl sodium and butyl
chloride;2 ...
1. Schramm, Ann. 218, 388 (1883).
2. Morton and Fallwell, Jr., J. Am. Chem. Soc. 60, 1429 (1938).
|
Taken from: http://www.orgsyn.org/orgsyn/prep.asp?prep=cv2p0047
=========================================
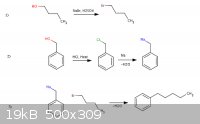
Preparation of n-propylbromide
This is blatant for anyone familiar with organic chemistry(imo) and comes from Org. Syn. No big deal there...
Preparation of Benzylsodium(maybe lithium?):
Benzyl Alcohol + HCl -> Benzyl Chloride (J F Norris, American Chemical Journal 38: 627-642 (1907) )
Here is where I get shakey(due to lack of information):
The preparation of benzyl sodium seems intuitive to a degree. Put a septum on an anhydrous flame dried flask, along with a drying tube packed with
CaCl2, slowly inject anhydrous benzyl chloride solution through the septum. The reaction should be exothermic and possibly require cooling(?). The rxn
should be possible at room temp(?). If there is remaining sodium metal, could it be quenched with methanol and the two layers separated(Benzyl Sodium
& methanol/trace NaOMe)?
The other problem I've had thinking about this is, is HCl formed? If so it would sort of 'poison' the Na, making NaCl..
I'd feel a lot more comfortable if I had a reference or some advice from the greater minds and hands here on this. I was also thinking, Lithium metal
might be a cheaper OTC alternative(benzyllithium), though I don't know enough about the actual chemistry to say whether or not this is an accurate
statement(pertaining the next reaction).
Preparation of n-amylbenzene:
If there is no remaining sodium metal from the end of reaction 2(slight excess benzylchloride) couldn't one inject the n-propyl bromide into the same
set-up?
I may be incorrectly assuming no solvents are used in these latter two steps, however, with two alkylating reagents I'm not sure what solvents would
be best to use.
================================
Keep in mind, this will all be small scale, grams, at most, I know better than to start big on new reactions. Not to mention my needs only require
2-3g of the final product anyways. I don't want to have any significant concentration of benzyl chloride or propyl bromide volatilized, even if I will
be working outside with a basic respirator, gloves and full goggles approved for chemistry.
Thanks sci. mad. sorry, to draw out a huge post on something probably trivial. Though I want to be precise, share my intentions, and show that I have
put in some effort here(researching/thinking).
[Edited on 11-8-2011 by smaerd]
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Quickest and cheapest would likely be to buy it. But if I was set on making it I would follow OrgSyn. Their procedures are published by experts and
checked (performed) by peers.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
The only problem I have with org. syn.'s procedure is namely, n-butyl p-toluenesulfonate. Which would not be a problem, if it didn't require,
p-toluenesulfonyl chloride. Which wouldn't be a problem if it didn't require SO2Cl2, or only available by the 500g quantity. Wish there were more
small suppliers. That procedure beats a wurtz fittig any day of the week. I must quit whining though, and find a solution.
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
You will not be able to touch the excess sodium with methanol. The NaBn will strip the acidic protons from it irreversibly much faster than it will
react with the solid sodium.
How about acylating benzene with pentanoyl chloride and AlCl3, then reducing by any of a number of reactions.
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
Chlorination of the acid to form the acyl chloride requires heavy reagents(SOCl2, PCl3)? War-gas, and things that use strictly watched elements  , Though that would be ideal! , Though that would be ideal!
Valerophenone is available to the amateur but its much more expensive(doubley so) than amylbenzene and still requires a clemmenson or a wolfkishner.
edit -
Ahh yes you are absolutely right about the methanol thing. Also the wurtz fittig should be performed in toluene apparently(not solventless as my OP
assumed). This is what they mean by an organic chemist must be able to consider their entire ''chemical environment'' with analytical thinking.
Learning new things is fun  , thank you for the suggestions so far. , thank you for the suggestions so far.
[Edited on 12-8-2011 by smaerd]
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
Actually hold the phone, I take back what I said about those crazy chlorinating reagents.
Edit oops - This journal has been posted already and is critical for this.
Reaction of carboxylic acids with tetrachlorosilane
http://www.sciencemadness.org/talk/files.php?pid=186775&...
Little nervous about generating Cl2 gas, but I have a pound of finely ground chromatography sillica(is this pure silica?). Yields are high enough,
considering how cheap amyl alcohol is.
Time to hit the drawing boards yet again. Even better yet acylation can use milder lewis acids than AlCl3 iirc. .
Thanks everyone sorry for changing my posts around like 50 times, I get a little too excited  /. /.
Edit -
No need to post the rxn scheme for this, anyone who knows very basic o. chem. reactions should be able to reason it out. When I post a megathread
though, about the initial project mentioned in the first few paragraphs of this thread I will likely include this preparation and my experimental
results along the way.
To everyone, thanks so much sci. mad. You've helped me tremendously over the past year or so. I don't believe I could feel more ready for Organic I,
next semester. I think I'm turning into a synthesis nut  . .
[Edited on 12-8-2011 by smaerd]
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
You need not even work with SiCl4. The anhydride will work fine as well and can be produced analogously to acetic anhydride as described elsewhere on
this forum. Do note that the pentanoic acid likely smells extremely foul.
[Edited on 8-12-11 by UnintentionalChaos]
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
Thank you for that warning! I will probably try and charge a drying tube with a weak base, and activated carbon to try and eliminate odors during the
oxidation(as well as working out-doors).
Good-tip on the anhydride, search function here we go!
edit - yea damn, SiCl4 is hard to prepare anyways. Passing dry Cl2 gas over 400*C, Si metal through a distillation flask with a drying tube, is far
from practical... I found maybe 300 posts about the preparation of acid anhydrides ... Back to researching.
[Edited on 12-8-2011 by smaerd]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by smaerd  | | So it is my intention to investigate the products(isomers) from the original Friedel Crafts reaction, amylchloride, benzene, and AlCl3(iirc). The goal
is to use old-school analytical techniques. In order to do so, I will have to develop standards, to compare products against. |
As far as I know, Friedel-Crafts alkylation of benzene with n-pentyl chloride should give no detectable traces of n-pentylbenzene. The products should
be 2-phenylpentane and 3-phenylpentane. Traces of rearranged products might also be present. Let us know if you ever obtain any GC evidence of
n-pentylbenzene formation. I guess that would be an interesting find.
| Quote: | | Org. syn. has a procedure for n-amylbenzene, however it uses a pretty rare chemical, that requires a rare chemical or three to make. So it is sadly
out of the question. |
If you consider butyl tosylate and benzyl chloride as rare chemicals, then you really should reconsider and try some other experiments first. Once you
get some experience you can re-reconsider and come back to this issue, but at the moment this is certainly above your level of lab skills.
Besides, benzyl chloride can couple with grignard reagents, so you could get away by the reverse reaction, coupling butylmagnesium bromide with benzyl
chloride. Kumada coupling of n-alkyl bromides with arylmagnesiums have been recently described in the literature, and even the inverse coupling if I remember correctly.
| Quote: | | After consulting with a couple chemical supplies, the lowest price I can find for n-amylbenzene was about 50$ for 25 grams of 98% purity(yellow
liquid? references say it should be colorless). I'm simply too cheap for this .
Why spend 50$ for an excess of a chemical when I can spend 30$, and have a couple useful left-over chemicals and the experience of preparing it?
|
You will hardly be able to make 25 g of this compound at a lower price than that, but I guess the experience, even if failing, is worth more than 50$.
Besides, if you are going to use it as a reference standard, you will have to follow exactly a published procedure or else you will have to fully
characterize the product, which means you will have to do 1H and 13C NMR, bp, MS, IR, GC purity...
| Quote: | | A few papers have helped to light the way, however I can't seem to find this one's abstract on any of the popular journals: Morton and Fallwell, Jr.,
J. Am. Chem. Soc. 60, 1429 (1938). |
Makes me wander if you even tried or you like it to be baby-sited all the way there... DOI: 10.1021/ja01273a046
But on a more serious note, if you even go as far as needing baby-sitting for locating articles, you surely can't expect us to advise you in an
organosodium reaction which will likely go wrong and may cause you serious injuries with painful burns!
Please, for the sake of your future, become responsible in what you do and start properly. You can't just jump into reactions that even people with
years of lab experience would be extremely reluctant to perform.
| Quote: | | Keep in mind, this will all be small scale, grams, at most, I know better than to start big on new reactions. Not to mention my needs only require
2-3g of the final product anyways. I don't want to have any significant concentration of benzyl chloride or propyl bromide volatilized, even if I will
be working outside with a basic respirator, gloves and full goggles approved for chemistry. |
If you only need 2-3 g then buy valerophenone and deoxygenate it by hydrogenation in methanol. You can do this at ambient pressure with a couple of
mol% of 5% Pd-C. It is safer, simple and as cheap is it can get. After you get TLC confirmation the reduction is over, all you need to do is filter
trough celite and rotavap. The product will be pure enough for use as a reference even without vacuum distillation and fractionation which is not
something that can easily be done on small scale. There is already a literature precedent, so you could do with only a bp and IR. Do this under
supervision of someone who has experience with hydrogenations ("safer" does not equal "safe"). Pd-C can ignite flames if thrown in a flammable solvent
in the presence of air.
It also appears to me that you are unaware that valeroyl chloride is commercially available and cheap. So, if you are going to investigate a
Friedel-Crafts alkylation on benzene you might as well practice first by doing a Friedel-Crafts acylation on it and obtain the required valerophenone
as you go.
| Quote: | | Thanks sci. mad. sorry, to draw out a huge post on something probably trivial. Though I want to be precise, share my intentions, and show that I have
put in some effort here(researching/thinking). |
That's all fine and great, but what I lack in your posts is at least a tiny bit of realism, proper rationality and concern for your own safety. You
can't fly before you learn to fly, and you don't learn by jumping from a skyscraper without a parachute. Also, you need to work on your article
reading habits. You should never draw conclusions about the experimental parts based on abstracts. Articles need to be read, particularly their
experimental part, before you even go to any vicinity of a lab, especially when dealing with such hazardous reactions.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
You could make valerophenone, via dry distillation of valeric acid/benzoic acid salts (similar to making acetophenone from benzoic and acetic acid),
then you could reduce the aryl ketone with Pd/c.
I doubt however you will make it for less than $50/25g.
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
@Nicodem - I totally agree that the original scheme was way over my head and hands. Doubt I would have died or anything trying it on such a small
scale, especially after obtaining the reference. I'm very grateful that an easy alternative is possible. Although...
I never considered benzyl chloride a rare chemical, and in-fact posted a reference to which it is very easily made(not to mention the many threads
pertaining it). I'm not new to gassing HCl. The butyl tosylate however is what I was referring to(in my second post). Sure I could have ordered a kg
of p-TsCl but it's not worth it, and I'm not made of money, though I have the proper equipment and know-how to prepare it(butyl tosylate not
p-TsCl)... Chlorination of p-TsOH is sketchy at home which is what I was referring to.
Actually in the references section I requested the article I couldn't find at the time of posting. Some hours later, and received it(from solo). Sorry
for not updating that part of the post. However, the acylation appeared far more attractive (as I posted a few posts down) once I found a means to
procure valeroyl chloride using easily attainable chemicals.
Now if you consider Pd/C cheap, and hydrogenations safe, well, I'm confused. I'm waiting to perform those in class before ever trying one at home. A
clemmensen is with-in my capabilities and I have the chemicals for it on hand(yes I know about mercury salt toxicities). Although the abstract of org.
syn. says the reduction can be performed with Zn and HCl alone.
Though valeroyl chloride may be cheaply available commercially. Though, like many others I can't order from laboratory supplies that do not ship
residentially, and would prefer not to order from china or whip up a phoney business. I think it would be fun to practice another primary alcohol
oxidation to carboxylic acid even if it's stinky. As well as repeating the SiCl4 chlorination in the paper I posted above(seems really easy), and I do
have a source for this chemical at a 'reasonable' price.
last edit sorry - To show you I'm not a complete and total lost dreamer here is the second scheme worked out. I know my style is 'juvenile' for
drawing these things, but I write them so I understand them.

@smuv - Valeric acid is pretty cheap to make, and titrations are safe . I just
wonder what all it would yield and how hard it would be to clean up. This would be the cheapest most low-tek route. I can't imagine the yields being
very high. Although I was planning to do something similar to obtain benzene. Maybe I'll try it out instead of all of this after a bit of research on
it. Sure beats SiCl4 or anhydride preparations.
I may not be able to beat the price of $50/25g, but like I have said in the initial post. I want experience, I don't need 25g, and 50$ can buy me
almost all of the chemicals I need to make 1g-2g that I need, with a large excess of useful chemicals to keep in the lab.
Again I really appreciate you guys looking out for me, and that is why I post here. My chemistry professor likes me and said if I ever wanted to
perform an experiment, I could discuss it with him and do it(as long as it isn't NI3, etc). So for scarier stuff, I will consult him. Again sorry for
a massive post.
[Edited on 12-8-2011 by smaerd]
|
|
|
mr.crow
National Hazard
Posts: 884
Registered: 9-9-2009
Location: Canada
Member Is Offline
Mood: 0xFF
|
|
I'm interested in this type of reaction too
FeCl3 is more easily available and more stable to storage than AlCl3 but has slightly lower yields. Anhydrous of course, not the stuff for etching
circuit boards.
You can make your pentanoic chloride by reacting with a less volatile acid chloride like benzoyl chloride. It would be easier/safer to make pentanoic
anhydride by heating the sodium salt with sodium pyrosulfate. Some members have had success making propionic anydride this way but not acetic.
pentanoic acid and friends are going to smell like a wolverine's asshole
Double, double toil and trouble; Fire burn, and caldron bubble
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
I would love to see descriptions like this in scientific papers. I might read a whole lot more of them.
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
@UnintentionalChaos - me too
@Mr.Crow - thanks for the tip. I have a pound or so of pyrosulfate laying around. The anhydride route seems most sensible as much as I hate to say it.
Salts of stinky acids are nice, anhydrides by nature are dry(redundant guy is redundant), it's cheaper than SiCl4, and surprisingly gives high yields
with the propionic salts.
Here's the pKa - 4.84 (Dissociation Constant at 20 C) taken from, http://chemicalland21.com/specialtychem/perchem/valeric%20ac...
@smuv - for the acetophenone synthesis yields are about 6%, probably worse for this compound, I don't like that. It was a good suggestion though. Back
to acylation .
[Edited on 12-8-2011 by smaerd]
|
|
|
Dr.Bob
International Hazard
Posts: 2659
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: No Mood
|
|
From wikipedia, where it is quoted from several scientific papers: (They do all but describe it in terms like the above in some other papers).
Their disagreeable odour is legendary. To quote from Lieke, "Es besitzt einen penetranten, höchst unangenehmen Geruch; das Oeffnen eines Gefässes
mit Cyanallyl reicht hin, die Luft eines Zimmers mehrere Tage lang zu verpesten, ..." (It has a penetrating, extremely unpleasant odour; the opening
of a flask of allyl [iso]cyanide is enough to foul up the air in a room for several days). Note that in Lieke's day, the difference between isocyanide
and nitrile was not fully appreciated.
Ivar Karl Ugi states that "The development of the chemistry of isocyanides has probably suffered ... through the characteristic odor of volatile
isonitriles, which has been described by Hofmann and Gautier as ‘highly specific, almost overpowering’, ‘horrible’, and ‘extremely
distressing’. It is true that many potential workers in this field have been turned away by the odour.”[4] Isocyanides have been investigated as
potential non-lethal weapons.[5]
Isonitriles make you want to vomit without being able to place the smell. Small, alkyl, ones are the worst.
Bob
|
|
|
|








