Megak
Harmless

Posts: 12
Registered: 8-12-2011
Member Is Offline
Mood: No Mood
|
|
Oxidation of Isopropanol
Hey everyone
I was thinking about this today and was wondering what reaction is the production of acetone. I'm looking more for the science of it, I'm uninterested
really in efficiency. So, I presume that
(CH3)2CHOH (l) + NaClO (l) -> (CH3)2CO
I doubt NaClO is an efficient oxidizer, but it's the first one off the top of my head. I also believe that this reaction wouldn't occur without a
catalyst, though I don't know what that catalyst is.
Any thoughts?
|
|
|
ThatchemistKid
Hazard to Others
Posts: 132
Registered: 2-6-2010
Member Is Offline
|
|
You will get oxidation of the alcohol to acetone with that reaction but it will not stop there you will then form chloroform and sodium acetate. It is
called a haloform reaction.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Megak  | Hey everyone
I was thinking about this today and was wondering what reaction is the production of acetone. I'm looking more for the science of it, I'm uninterested
really in efficiency. So, I presume that
(CH3)2CHOH (l) + NaClO (l) -> (CH3)2CO
I doubt NaClO is an efficient oxidizer, but it's the first one off the top of my head. I also believe that this reaction wouldn't occur without a
catalyst, though I don't know what that catalyst is.
Any thoughts? |
Secondary alcohols can be oxidised to ketones by means of potassium dichromate (possibly also potassium permanganate): IPA to acetone, for instance.
But that's not the industrial method of producing propanone...
A bit of background:
http://www.chemguide.co.uk/organicprops/alcohols/oxidation.h...
[Edited on 9-12-2011 by blogfast25]
|
|
|
JibbyDee
Harmless
Posts: 38
Registered: 25-11-2011
Member Is Offline
Mood: No Mood
|
|
Yeah this is an easy method of producing chloroform. I'd opt for permanganate as the oxidizer instead. If your aim is to explore simple organic
reactions like this then you might as well go ahead and test out the ClO oxidation of IPA. Chloroform is a very useful solvent so knowing how to
produce it is a good skill for the home chemist. Haven't actually made any myself, but once I get some decent reagent bottles to store the chloroform
in, I'm going to try this reaction. I distilled about a 500 mL of dichloromethane last year for my lab but I stored it in a glass canning jar and a
few weeks later when I went to get some of it, it was gone lol. Solvents like these get through the tiniest gaps so you really need proper containers
to store them in. In some oxidation reactions (such as the permanganate oxidation of ethanol to produce acetaldehyde and ultimately acetic acid) you
can distill the product before it is further oxidized into the next compound, maybe you can distill off the acetone before it is further oxidized into
chloroform. IPA has a boiling point of around 80C whereas acetone has a BP of around 60C.
[Edited on 10-12-2011 by JibbyDee]
|
|
|
Megak
Harmless
Posts: 12
Registered: 8-12-2011
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by JibbyDee | Yeah this is an easy method of producing chloroform. I'd opt for permanganate as the oxidizer instead. If your aim is to explore simple organic
reactions like this then you might as well go ahead and test out the ClO oxidation of IPA. Chloroform is a very useful solvent so knowing how to
produce it is a good skill for the home chemist. Haven't actually made any myself, but once I get some decent reagent bottles to store the chloroform
in, I'm going to try this reaction. I distilled about a 500 mL of dichloromethane last year for my lab but I stored it in a glass canning jar and a
few weeks later when I went to get some of it, it was gone lol. Solvents like these get through the tiniest gaps so you really need proper containers
to store them in. In some oxidation reactions (such as the permanganate oxidation of ethanol to produce acetaldehyde and ultimately acetic acid) you
can distill the product before it is further oxidized into the next compound, maybe you can distill off the acetone before it is further oxidized into
chloroform. IPA has a boiling point of around 80C whereas acetone has a BP of around 60C.
[Edited on 10-12-2011 by JibbyDee] |
Hmm..... I was thinking about this and if I added the NaClO slowly and have the IPA in excess instead, I should limit my chloroform production, and
even then i can pump it into an ice bath to condense it.
Though, I think if you want chloroform, just go straight with acetone and bleach, you'll get better yields. And from what I remember about chloroform,
i think you can stop some of the evaporation by putting it in cotton balls
|
|
|
Megak
Harmless
Posts: 12
Registered: 8-12-2011
Member Is Offline
Mood: No Mood
|
|
So, I did try this and I didn't play around with it much, but after heating the mix up, I did notice some chloroform mixed with water.
Where from there? I have no idea
EDIT: Ok, so I think I know what has happened. My solution has oxidized a great amount (completely colorless) after some time. When I burned the
solution off, I burned off the alcohol and acetone, bleach remained with chloroform. That's why it smelled sweet, I think
[Edited on 12-12-2011 by Megak]
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
How much potassium or sodium dichromate or Potassium permanganate is needed for oxidation 1 lit of Isopropanol to acetone?( i like to try it)
I think passing Isopropanol from Hot Copper Tube is also possible route for oxidation of Isopropanol to acetone
[Edited on 21-2-2012 by Waffles SS]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
I was trying to oxidize IPA via catalytic oxidation on CuO/MgO catalyst described in the article below. As you can read there, the pure CuO catalyst
has at least 10 times lower activity at the temperatures I use (<400C to avoid potential explosions because of poor temperature control).
First I tried to perform conversion using air as a carrier for IPA, by bubbling the air through a slightly heated IPA and passing the resulting air
into a glass tube, containing the catalyst hold between two pieces of glass wool; on the other side of the tube I placed a condencer. After few hours
I've got some liquid that doesn't give any reaction with sodium bisulfite (clear solution), so most likely there's no acetone formed.
Besically I've got an oxidation conversion and the catalyst was strongly overheated glowing red (famous oxidation of acetone with copper penny or rod
to methane, ketene and CO2), but not the dehydration to acetone I need.
Next time i tried performing this conversion without any carrier, using sole IPA vapour by boiling the alcohol-water azeotrope. And again I got the
liquid that gives almost no signs of acetone in it -slight turbidity in the bisulfite test and no definite red coloration in the nitroprusside test.
It may be traces of acetone though.
Both times I had 3 g of catalyst with ratio CuO:MgO = 2:8 mole; reactor temperature of 200-300 C. I didn't measure the gas flow, but I can tell that
it took 1-2 hours to pass 200 ml of IPA through the tube in the second experiment (so basically it's 1.5 mole/hour flow for 3 g of catalyst). The tube
internal diameter is 15 mm.
I'm doing such flow reactor catalytic conversions first time and I don't know much about them, so could anyone pinpoint my mistakes?
I don't have a source of the nitrogen neither the argon, so I can't use them as dilluents in the conversion. Maybe I just missinterpreted the article
and in fact you can't get any decent yields using this catalyst, can you? Maybe a relatively low catalyst activity is a problem? AFAIK it lays
somewhere at 0.01 mole/h/g (0.01 mole per hour for 1 g of catalyst) , so 3 g of the catalyst should convert 2 g of IPA per hour using a perfect setup,
right? So to make the reaction viable I need like 1 kg of catalyst packed into a lots of tubes. Or maybe one long tube packed with 100 g of catalyst
capable of converting 50 ml of IPA per hour.
I might try to isolate some acetone from my last mixture via distillation but I don't think I will succeed.
Attachment: a014.pdf (528kB)
This file has been downloaded 1416 times
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
I'm feeling lucky today. Found a good old patent that describes a similar CuO-MgO catalyst, but they use CuO:MgO = 6:4 and their target compounds are
methyl isobutyl ketone and diisobutyl ketone, though theirs catalyst starts to give acetone after few days of operation.
So, I actually already pointed my problem in my previous post: acording to the patent this catalyst is capable of converting 100 grams of IPA per hour
per 1KG of catalyst. Although the guys in the patent just mixed the CuO and MgO instead of decomposing Cu(NO3)2+MgCO3 mixture, so probably I could get
200-400 g/h/kg rate with the improved catalyst.
I'm just thinking about what reactor I should build for that, because the tube is just too small for that amount of catalyst. Iron will probably be
oxidized in contact with copper salts, glass tube needs to be of really big diameter and it might get cracked from because of the thermal shock and
quartz tube of a big diameter costs a lot, ceramic has the same thermal shock problems as a pyrex glass. It looks like copper tube will be the best
choice. Or probably some high flask immersed into Ca-K-Na-NO3 salt.
Attachment: Process for preparing ketones (isopropanol dehydration) - US2891095.pdf (464kB)
This file has been downloaded 492 times
[Edited on 8-4-2015 by byko3y]
|
|
|
Jimmymajesty
Hazard to Others
Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
Hi byko3y
Try going with copper tube, or rather brass tube if you have it.
I do not think that it would worth the hassle to prepare metal oxid catalyst mixtures. Just pack your brass tube with copper wire and heat it up in a
controlled way.
If you use air to oxidize the Ipr-OH do not pass the resultant liquid over and over again since you will only oxidize your acetone, you can however
reflux the products over the catalyst -if no air is let into the system- you only go till acetone this way note however that acetone is more volatile
than Ipr-OH so you only wasting time after the equlibrium is reached.
|
|
|
macckone
International Hazard
Posts: 2159
Registered: 1-3-2013
Location: Over a mile high
Member Is Offline
Mood: Electrical
|
|
Key trick in this reaction. Get as close to 400C as possible without going over. At 300C the reaction is going to be so slow as to be almost
nonexistent. Once you get up to 375C the reaction should be going pretty good. This is similar to making formaldehyde from methanol. Going over
400C is going to ruin your catalyst so you don't want to do that. The exact temperature is going to depend on the exact catalyst. Copper gauze is a
nice substance to use as a catalyst for this type of reaction and less sensitive than the CuO/MgO catalyst mentioned. As mentioned just heat the
gauze in a stream of air at 375C.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
The conversion of copper oxides below 120°C is negligible. Supported copper nitrate can do the job in reflux, but the yield is not as good as you
would expect from such costly reagent.
Looks like you didn't look inside the first article I posted, here's the picture from it:
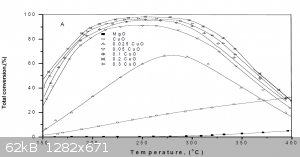
Figure (6): Total conversion of iso-propanol as a function of reaction temperature on CuO/MgO solid precalcined at 400°C using various molar ratios
of CuO.
As you can see, pure CuO-MgO is 10 times more powerfull catalyst then a pure CuO at 200-250°C. Also, copper wire has little sufrace area, compared to
an order of square meter per 1 g of catalyst.
And I don't think there's possible any conversion of IPA to acetone until you get rid of oxygen completely. AFAIK, at O2:IPA=8:1 oxidation peaks it's
maximum. That basically means you get no acetone in that case.
And I don't like to compera industrial methods to home-lab methods, because I can't afford high pressures and temperatures. Although, I have no
detailed decription of any industrial method of IPA oxidation on copper oxide.
I hope macckone knows he says, but I would like to read some article or book about that. I'm totaly agree that copper catalyst will be destroyed at
temperatures higher than 400°C, also at such a high temperature IPA or acetone will either decompose or catch fire even without any catalyst.
[Edited on 12-4-2015 by byko3y]
|
|
|
macckone
International Hazard
Posts: 2159
Registered: 1-3-2013
Location: Over a mile high
Member Is Offline
Mood: Electrical
|
|
Here is another paper on decomposition of isopropanol over MnO. After some research, I found that the decomposition of acetone isn't a major concern
at the temperatures we are discussing. Obviously catalyst can lower that temperature. The dehydration is an obvious competing reaction to
dehydrogenation and has a much greater effect.
Attachment: v71-631.pdf (323kB)
This file has been downloaded 936 times
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
"High-purity nitrogen at atmospheric pressure was used as a carrier gas and the catalyst bed temperature was controlled to + 0.1" from 300 to 400 "C"
:/
I have a link to an article about oxidation of IPA with air, but I could not find a full document. http://pubs.acs.org/doi/abs/10.1021/ie00072a007
|
|
|
Jimmymajesty
Hazard to Others
Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
That is indeed a nice graph, so how about...
1,oxidizing some copper as an anode with KNO3 electrolyte
2, filter off the CuO.
3,Buy some MgO from a pharmacy
4,Mix the two oxide, wet them with water,
5,Spiral up a copper tubing of 1cm dia,
6,Let the MgO CuO paste into the tube, put the spiral between two hotplates
7,Set the hotplates to max to dry the catalyst
8,Attach a dropping funnel and an aquarium pump to the inlet,
9,Collect the acetone with a condenser at the end,
10,Post your success
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
What I did was:
CuSO4 + NaOH -> NaSO4 + Cu(OH)2 --boil--> filter off CuO
CuO + HNO3 -> Cu(NO3)2 + H2O (I used a solution, the crystals are hygroscopic and you need to mix salts in the solution anyway)
MgCl2 + 2NaHCO3 → Mg(HCO3)2 + 2NaCl -- bubble air --> MgCO3 precipitates, filter of.
Now mix Cu(NO3)2 and MgCO3 as needed. In fact you can use basic magnesium carbonate ( Mg(OH2)*3MgCO3 ), and probably even Mg(NO3)2, but I tried to
stick the procedure in the article.
And no, I failed, as I posted above. I got back 99% of IPA I passed through the tube. Some white cloudy oil left in the flask.
|
|
|
macckone
International Hazard
Posts: 2159
Registered: 1-3-2013
Location: Over a mile high
Member Is Offline
Mood: Electrical
|
|
Try sci-hub.org and type in the doi: 10.1021/ie00072a007
My experience is that copper gauze is the easiest for the home chemist.
The yield may only be 30% but you can recycle the isopropyl alcohol.
With formaldehyde it is a bit easier because the formaldehyde polymerizes
and doesn't distill.
You could try the following:
Get a 1" copper tube with loosely packed copper gauze.
Fill with a solution of magnesium chloride and copper chloride.
Then add sodium carbonate solution to the tube.
This should make the mixed carbonate precipitate on the copper gauze.
Drain off excess solution.
Then heat to 400C to decompose your carbonates.
Repeat as many times as necessary to get the proper fill.
This should give a higher surface area and a well supported catalyst.
Now for the mechanical part, the 1" copper tube is going to need to attach to something. At these temperatures you should probably sweat on a
threaded coupling before filling the tube. The threaded coupling can have what ever you need screwed into it. In this case it is probably going to
be a fitting leading to your metal boiling container and another to your condenser which should probably also be metal.
I suggest metal because if something goes wrong metal is less dangerous that glass shrapnel. Not that it can't kill you but you have a couple of
holes instead of hundreds for the doctors to work on. In other words this isn't safe don't try this at home. Unless you are a mad scientist and then
just don't blame me. You have been warned.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
sci-hub did the job.
Indeed, I was right, the major product for high O2:IPA ratio is CO2.
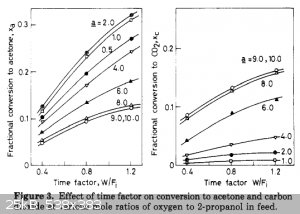
a = O2/IPA molar fraction; W/Fi = catalyst weight / flow rate = g / (mol/h) = g*h/mol
So for a 10 g of CuO:ThO:pumice catalyst, time factor of 1.0 g*h/mol, O2/IPA = 1-2 (or mass ratio 0.5-1.0) at 300°C you can convert 10 mole (700 g or
1 liter) of IPA to 200 g of acetone per hour. Which is pretty decent. Although I have no thorium available.
The paper says the drop of the reactor temperature from 285°C to 240°C leads to a drop of the conversion rate 10 times.
Also, conversion graph seems to become almost flat somewhere at 5+ g*h/mole, so I should not use higher flow rate if I want good yields. And I used
time factor W/Fi = 2 g*h/mole in my last experiment, which lower yields even for CuO:ThO:pumice at 300°C, and I'm sure at 200-250°C the CuO:MgO
catalyst is capable of processing few times less IPA flow (but giving really high conversion).
|
|
|
Jimmymajesty
Hazard to Others
Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
I did a run today.
-the IPA was brought to boil, then the wattage on the kanthal wire was set so that slight propylene generation could be observed. The wire glowed in
the dark but it took time to notice.
-let air in below the wire at 5ml/sec, let the setup do its job for 1,5hr
-closed the air inlet and started to fractionate the mixture.
Problem is I could not get the top thermometer below 73°C even with infinite reflux, which is strange because the sample from the top quickly
evaporated on the hand and left very little IPA stain.
Bottom line is, this can be easily done by the setup posted several replies above. Heat the IPA till propylene formation then set the temp back a
little, let some air in and run the condensate through the tube several times, then fractionate the mixture in a separate setup.

|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Okay, folks, now you can commit suicide, because actually the OP's reaction works
(CH3)2CHOH + NaClO -> (CH3)2CO + NaCl + H2O
Student preparation of acetone from 2-propanol
J. M. Kauffman and J. R. McKee
J. Chem. Educ., 1982, 59 (10), p 862
DOI: 10.1021/ed059p862
Attachment: Student preparation of acetone from 2-propanol.pdf (843kB)
This file has been downloaded 650 times
Although the catalytic way is cheaper, it's waaaay more time consuming, until you want to make 10+ liters of acetone.
|
|
|
Jimmymajesty
Hazard to Others
Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
byko3y I thought you were after an efficient catalyst, if your goal is to get 10+ liters of acetone the most simple action is to buy it online, I
could even order a 100l drum if I wanted to from synthetic resin supplyers and it is also as cheap as water.
The catalytic way not just cheaper but also high yielding so you do not need to worry about side products. I will repeat the experiment and post the
results as soon as the acetone will be banned.
[Edited on 24-4-2015 by Jimmymajesty]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Jimmy, in my country I can freely order online up to 10 year in jail for contraband of a large amount of acetone. And if they find acetone peroxide in
it - I might get charged for terrorism.
I'm just getting surprised over and over again by people who see a lot of someone's afford to prepare some compound, and giving advices like "if your
goal is to get 2+ liter of safrole the most simple action is to buy it online, I could even order a 10 liter bottle of it" or "I have here a 1 kg of
LAH on my shelf, I see no reason why you would need to use sodium dithionite instead".
Moreover, here acetone is as cheap as 5$ per liter of 99% pure, in case you are an organization holding a license to handle drugs precursors.
[Edited on 24-4-2015 by byko3y]
|
|
|
Mesa
Hazard to Others
Posts: 264
Registered: 2-7-2013
Member Is Offline
Mood: No Mood
|
|
http://jcsp.org.pk/ArticleUpload/51-281-1-CE.pdf
It doesn't fit the true definition of a catalyst as far as oxidation of the alcohol goes but it is an interesting catalytic system. Costs next to
nothing and proceeds at room temperature too.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Actually, there's an example of aliphatic alcohol oxidation. And it's basically an oxidation by hypochlorite, catlyzed by nickel hydroxide. http://pubs.acs.org/doi/abs/10.1021/jo0612574
I'm not sure if the nickel compounds plays any role there, because you can oxidize benzyl alcohol and isopropanol with hypochlorite without the
nickel.
PS: and actually I've already tried this method, but failed to isolate any acetone.
[Edited on 24-4-2015 by byko3y]
|
|
|








