Asphyxiated
Harmless

Posts: 20
Registered: 29-2-2012
Member Is Offline
Mood: No Mood
|
|
First attempt at the rearrangement of 2-phenylpropanal
Hey there,
I posted on here a couple weeks ago about the possible rearrangement of 2-phenylpropanal (2-phenylpropionaldehyde) to phenyl-2-propanone. I was using
this method:
| Quote: |
2-Phenylpropanal can be rearranged with either mercuric chloride (HgCl2) or sulfuric acid (H2SO4) to form the isomeric phenyl-2-Propanone (P2P).
2-Phenyl-propanal (hydratropic aldehyde) is used in the perfume industry. 2-phenylpropanal can also be made from alpha-methylstyrene.
The CAS number for 2-phenyl-propanal is [93-53-8], and synonyms for it include Hydratropic aldehyde; 2-Phenylpropionaldehyde; Cumenealdehyde;
alpha-methyl benzeneacetaldehyde and alpha-methyl phenylacetaldehyde. Boiling point 92-94°C/12mmHg, 222°C/760mmHg.
There are other ways of performing this rearrangement, 2-phenylpropanal is isomerized to phenyl-2-propanone in up to 87% yield by passing its vapor
over an iron zeolite catalyst bed at 500°C, followed by condensation of the vapors and redistilling the P2P.
Even if the method below which uses mercuric chloride is higher yielding than the one using cold sulfuric acid, I would definitely reccommend the one
with sulfuric acid, as it is much cheaper to use, and is not disastrous for your health or the environment. 60g of mercuric chloride contains 45 grams
of mercury, enough to poison a medium-sized lake if released into the environment, and if you happen to ingest it yourself, it will accumulate in your
body.
It is not possible to effectively separate 2-phenylpropanal (bp 222°C/760mmHg) from phenyl-2-propanone (bp 214°C/760mmHg) through simple
distillation and certainly not via vacuum distillation as the boiling points are too close. Fractional distillation could theoretically be used to
separate them, but the size of the column that would have to be used makes that option impractical. A good idea for separating a mixture of the two is
to oxidize the mixture with a mild oxidant which won't affect the P2P, but which will oxidize the aldehyde to 2-phenylpropionic acid. The acid can
then be separated from the ketone by dissolving the mixture in a non-polar solvent and washing the solution with dilute sodium hydroxide. The P2P
stays in the organic layer, which is then dried over MgSO4, the solvent removed under vacuum and the residue vacuum distilled to give pure P2P.
Method A
30g of 2-phenylpropanal is heated together with a mixture of 60g mercuric chloride (HgCl2, 1 eq.) and 450ml 75% ethanol in a pressure-safe sealed
glass container for 4.5h at 100°C in a boiling water bath, during which time a precipitate forms. Water is added, and the solution is steam-distilled
(during which operation the precipitate redissolves). The distillate is extracted with ether, dried, and the solvent is evaporated. The oily residue
is then vacuum distilled with a fractionating column to collect the phenyl-2-propanone in a yield of 80% or more, bp 92-101°C at 14mmHg. When 0.1
equivalents of HgCl2 was used, only 10% phenyl-2-propanone was formed, the rest consisted of unchanged aldehyde.
Method B
9 g of 2-phenylpropanal is slowly added with good stirring during 35 minutes to 40ml concentrated sulfuric acid, while the temperature of the reaction
mixture is kept at -16°C. After all the 2-phenylpropanal has been added, the mixture is allowed to stand at the same temperature for another 15
minutes, and then the mixture is poured onto crushed ice (100-150g is probably a suitable amount). When the ice has melted, the organics are extracted
from the water phase by 3x50ml diethyl ether, the pooled organic phases dried over MgSO4, the ether distilled off and finally the residue is vacuum
distilled (bp 91-96°C at 11 mmHg) to give 5.6g (62%) of phenyl-2-propanone. |
I went about the procedure using Method B as I had everything required (actually I have the HgCl2 as well but its expensive and didnt want to use 60
grams of it up just to see if this worked). I have not completed the experiment entirely but far enough to ask some questions.
I decided to double the quantities used in method B, due to the yield being only 5.6 grams (which would result in roughly 5.6ml of product, not a
lot). So I started by cooling a saturated NaCl solution to -16--18 C and then adding 80 ml of conc (98%) sulfuric acid to a 250ml RBF and allowing it
to reach -16C as well.
I as I waited I measured out 18ml of 2-phenylpropanal and began adding it slowly over 35 minutes. (Question 1: Should I have doubled the addition time
because I doubled the reagents being used?). The brownish colored liquid that was originally 2-phenylpropanal became a very viscous liquid when
contacting the H2SO4, much like it was polymerizing and became black, it then disassociated from itself and dissolved into the acid. By this I mean
that as it contacted the sulfuric acid the droplet more or less stayed together as it seemed to become much more viscous and also turned deep black
and then went into solution with the sulfuric acid, turning the entire mixture black rather quickly. The entire solution was black after only 3-5 ml
of 2-phenylpropanal had been added.
After adding the entire portion of 2-phenylpropanal I allowed the mixture to stand (which I assumed meant stop the stirring) at -16C for 15 minutes (
should this have been 30?) and then dumped it on to 225 grams of crushed ice. As the ice melted the solution seemed to be bi-phasic, there was a much
lighter lower layer and a thick black upper layer. After the ice melted the bottom of the RBF had to have been at least 50F if not hotter. I assume
that it is at this point that the remaining 2-phenylpropanal was converted to 2-phenylpropionic acid as hot sulfuric acid is an oxidizer and the
remaining 2-phenylpropanal must be converted to the acid in order to seperate according to the literature referenced.
I then dumped this entire solution into a 1000ml sep funnel and added 100ml of dry diethyl ether. Initially the the floated on top of the aqueous
phase, as expected, but after shaking (not much at all in order to avoid emulsions) it did not readily separate. I decided to leave it for a few
minutes ( 15 in total) to see if it would separate again. After 15 minutes there was a very distinct top dark black layer but the volume of this layer
was significantly less than the 100ml of ether I originally added to the solution. I decided to separate this layer either way and continue with the
extraction.
The first top layer smells rich in ether, which gives me some hope that it did actually seperate somewhat. After the separation I re-added the aqueous
phase to the sep funnel and added another 100ml of ether and shook gently. This time it did not separate out at all. I then left it for over 2 hours
to see if any separation would occur, it did not. (So next question, why would it not separate back out??)
I added yet another 100ml of ether to the solution, which sat on top of the aqueous layer and comes back with VERY gentle shaking. I have not shaken
it as much as last time in fear that it will all disappear again, and I am out of ether. I have roughly 50ml of a dark oily substance with a rich
smell of ether separated though. I plan to distill off the ether and then vacuum distill the residue to see if anything comes over in the right range.
Another strange thing that happened is that after the original extraction and pouring the solution back into the sep funnel the aqueous layer crashed
out a TON of crystals. I assume that these crystals are the carboxylic acid of the 2-phenylpropanal but I don't know why they crashed out of solution.
The ph of the solution didn't change from ether washes and I only added ether to it, nothing else. The temperature also dropped in the work
environment when this happened but warming the liquid back does not cause the crystals to go back into solution.
Perhaps someone could give some insights as to whether these crystals are the acid I think they are and maybe why they crashed out of solution. Or
address any other questions I laid out in the write up including with respect to the times I used in this experiment and also where did my ether go?
I plan to redo this experiment either tomorrow or the next day and use DCM as my organic solvent this time and see if it also disappears. DCM is much
more easily available to me than ether is.
Any way, Thanks in advance!
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
It's very nice to actually see some real experimentation and not only theoretical mumbo-jumbo. But I hope you have a high frustration tolerance,
because this might not be the easiest thing to do without proper analytical equipment.
It's no wonder that mixing organics with H2SO4 gives a mess. A famous beginner experiment is to apply conc. H2SO4 to sugar - there's a reason that
sugars are called CARBOhydrates.
I don't think you'll have necessarily to double the addition time since you also have also double the reaction volume. But mind that the reaction
volume goes linear your scale whereas the surface goes only with power 2/3. Meaning that removing heat becomes more and more difficult. So what you
will have to control closely is the internal temperature of your reaction.
The second thing you probably should take care of is adequate stirring. As in as powerful as possible. The way you describe things, I'm not sure that
it was.
I'm not sure that your dilute H2SO4 would quantitatively oxidize the aldehyde to the acid. Dil. H2SO4 is not really a powerful oxidizer. Why not do
some experiments on neat aldehyde first? It should be quite easy to separate acid from aldehyde by means of acid-base extraction. Of course, first hit
the literature on proven procedures. Phenylacetone is not the stablest substance there is, so there is some risk of destroying it with strong
oxidizers.
I faintly remember some discussion of using bisulfite adduct to separate the two. Maybe this helps in finding information.
Concerning the ether: Well, it does have an appreciable solubility in water, there is always some emulsion going on and some of it evaporates. What
you observed is to be expected. Use more ether , salt it out by adding NaCl/brine. Brine may also improve separation due to larger difference of
densities. But it can also give a mess.
As to your crystals: isolate them, dry them and do some caveman analytics: melting point, solubility in organics (with/without heat), solubility in
neutral, basic or aqueous medium (with/without heat). That should give you some ideas.
PS: I'm not an organiker - take everything with a huge grain of salt.
PPS: Good luck. 
|
|
|
Asphyxiated
Harmless
Posts: 20
Registered: 29-2-2012
Member Is Offline
Mood: No Mood
|
|
Hey, thanks for the reply.
I fixed the first mess by adding a total of 100ml of 10% NaOH solution. One of my chemist friends said that it was possible that the phenylpropionic
acid may have lost the oh group and causing it to crash out of solution so I first added about 25 ml of 10% NaOH solution and the crystals started
going back into solution. After adding a total of 100ml everything was back into solution and low and behold my ether layer came back as well. I am
not sure if this is due to the addition of NaOH or because that addition raised the tempurature considerably. (from roughly 20C to 30C), any how I
quickly extracted the ether layer and combined it with the prior top layer that I had extracted earlier. My chemist friend then wanted to re-do the
reaction with him present this time so we did.
I followed the same procedure except added the 2-phenylpropanal much slower this time than last time. 18ml was added over 50 minutes instead of 35. I
also used DCM instead of ether (as I was out of ether) and it seems to be going quite well. The first DCM wash easily separated and took the black
oily layer with it. On second wash with DCM even more black oily substance was extracted, much more than the first time. It is still separating as I
type this. I will then distill both the ether containing flask and the DCM containing flask to to compare results. I am assuming that this second
attempt with DCM is going to give me more yield, just because things went much smoother this time around. I haven't had to adjust the ph of the
solution yet. Although the sulfuric acid did freeze up on me this time at around -18C so i added water drop wise to lower the concentration to a point
where the freezing point would be below -18C, I added maybe 1-3 ml of water and everything was liquid again.
As for the stirring, it was maximum the entire time last time as it was this time. And if the H2SO4 does not oxidize the left over 2-phenylpropanal to
its acid then what does? Supposedly you need to oxidize the unreacted portion so that it can be separated via polar/non-polar solvents. The acid being
highly soluble in water and the p2p being highly soluble in organic solvents.
Anyhow, will report back on the findings from both flasks.
Cheers!
[Edited on 3/29/2012 by Asphyxiated]
|
|
|
Asphyxiated
Harmless
Posts: 20
Registered: 29-2-2012
Member Is Offline
Mood: No Mood
|
|
After distilling the first trial i only received roughly 5 ml of a clear oily substance in the receiving flask. I am not sure exactly what this
substance is, I will have to do some work up on it, but there isn't much to work with. It came over at roughly 75 C for vapor temp, which is way too
low unless my aspirator pulls a vacuum of more than 14mmHg (a joke) because the boiling point of p2p at 14mmHg is roughly 91-96C, however my oil bath
did reach over 160C so if there was any p2p in the first batch it should have came over... however I had a bad feeling about that run any way. It will
be much more interesting to see what is in the second batch tomorrow, now that I got my vacuum all figured out.
If anyone has any idea of what this substance is that I collected then feel free to chime in, because I have no idea. I believe p2p is suppose to have
a greenish hue to it but there may be so little of it in the vessel that any color is not noticeable. ANYWAY, for those that are interested, I will
keep a log here of my journey. I have plenty of 2-phenylpropanal to keep trying this out, so I think I will put this recipe to bed one way or another.
Either it works and will be documented here or it does not work and it will so be documented here.
Until then, cheers!
EDIT: However I would like to say that after this clear oily substance came over at ~75C there were no more vapors. There is a lot of black oily stuff
left in the boiling flask but it did not have a BP of less than 160C even at reasonable vacuum (if i had to guess maybe 100-80 mmHg). So if this clear
oily substance is not p2p then this first reaction was a failure.
[Edited on 3/30/2012 by Asphyxiated]
|
|
|
haroldramis
Harmless
Posts: 21
Registered: 6-1-2012
Member Is Offline
Mood: No Mood
|
|
I second that turd. This site is so festering with mental masterbation that when I see someone who does ACTUAL chemistry and then shares it with
everyone it surprises the hell out of me. A bright ray of sun poking thru the clouds of hypothetical nonsense.
|
|
|
Asphyxiated
Harmless
Posts: 20
Registered: 29-2-2012
Member Is Offline
Mood: No Mood
|
|
Second attempt yeilds p2p!
So I did the work up on the dcm extracts I did yesterday on my second run at the rearrangement of 2-phenylpropanal to phenyl-2-propanone and I am,
lets say, 80% sure that it worked this time. Not really in yeilds I was hoping for but I was able to obtain about 12.5 ml of a yellow oily substance
with bp 106-111C (at an unknown vacuum pressure, if this is p2p then that would correspond to roughly 20mmHg).
Some more experiments need to be ran to see if this is reproducible, I am not sure if any one else has tried this reaction besides the original paper
from 1927 but it is starting to look good.
Since p2p has a density of 1.003 g/mL then its weight is pretty much equal to its volume, so I received ~12.5 g from ~18 g of 2-phenylpropanal (18 mL
was used in the second run and 2-phenylpropanal has a density of 1.002g/mL). So i guess that is actually a decent yield. Going from the yield they
gave as 62% I got a respectable yield of 69%, even doubling the reagents, without knowing how that would truly affect the reaction.
I'd say that this synthesis is looking more favorable by the day, I had my doubts originally about whether it would work or not but it seems to work,
even though all of the chemists I know can't tell me how the reaction actually works or what happens where and why, it seems like some kind of magic
honestly. (I mean I know it isn't "magic" but its such a bizarre synthesis route that no one that I know that works/studied in the field of chemistry
can explain how or why this works.
Anyway, on to the pictures!! I only took a picture of the final product, I didn't really plan on documenting this originally so I didn't take any
other pics and the pictures don't do the color of the liquid justice. It's much brighter yellow/green, almost a neon color.
I am also looking for ways that the amateur can determine what substance I have with more exact'ness' than I have right now. The bp seems in the right
area but that does mean anything. The color is right, so thats good and the smell is right as well, but definitely not an NMR or MS/GC analysis by any
stretch.
Keep on cookin my fellow chemistry enthusiasts!
  
[Edited on 3/31/2012 by Asphyxiated]
[Edited on 3/31/2012 by Asphyxiated]
|
|
|
Chordate
Hazard to Others
Posts: 108
Registered: 23-2-2011
Member Is Offline
Mood: No Mood
|
|
Without GC/MS you are largely limited to wet tests. If you can get your hands on a derivatizing agent's that'd get you a way to get a sharp melting
point and help confirm identity. Dinitrophenylhydrazine might be tricky to acquire but derivatizing with it is easy and takes but a few minutes of
work, and it gives sharp melting points.
Otherwise the tests that come to mind here are schiffs, and the haloform test. Schiff's is considered hobbyist experiment so kits are quite easy to
acquire through the post. That'll tell you at least if you got the aldehyde out of your mixture and possibly give you some sort of qualitative
information as to purity.
The haloform reaction can be performed quite easily. Lugol's 5 % solution is pretty much pre-made haloform reagent, don't try and use tincture as the
ethanol can give you false positives. The hardest thing may well be that your material is insoluble in water, thus making it hard to do the test
without an appropriate solvent, usually a small amount of dioxane or glyme followed by dilution with water and performing the test. Control with
acetone until you figure out your ideal concentrations.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Asphyxiated  | Hey there,
I posted on here a couple weeks ago about the possible rearrangement of 2-phenylpropanal (2-phenylpropionaldehyde) to phenyl-2-propanone. I was using
this method:
| Quote: |
2-Phenylpropanal can be rearranged with either mercuric chloride (HgCl2) or sulfuric acid (H2SO4) to form the isomeric phenyl-2-Propanone (P2P).
2-Phenyl-propanal (hydratropic aldehyde) is used in the perfume industry. 2-phenylpropanal can also be made from alpha-methylstyrene.
... |
|
I will provide the reference to that quote in order to alleviate the original poster's transparent attempt at plagiarism:
http://www.erowid.org/archive/rhodium/chemistry/phenylaceton...
The original reference as cited there is Chem. Ber. 1927, 60, 1050.
Asphyxiated, I warned you before to cite sources. This is a science forum and we do not tolerate such behavior. Also, this is already the third thread
you open on the same topic and you did not even bother to link to the previous merged two. Please read the forum posting guidelines.
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
cause for celebration

Give me librium or give me meth!
Patrick Henry....
|
|
|
Asphyxiated
Harmless
Posts: 20
Registered: 29-2-2012
Member Is Offline
Mood: No Mood
|
|
Such a small forum I thought that any interested parties would know about my previous post, or as I stated "I posted a couple weeks ago about...." I
also don't see how I am attempting plagiarism, I did state that the paper was originally written in 1927, which would make it probably not mine. I'm
sorry that these things were not spelled out, I will do so in the future because the rules ask me to.
Maybe I am missing something here but isn't it at least kind of cool that the rearrangement seemed to work?
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
cool reaction. shitty plague product.
ʃ Ψ*Ψ
Keepin' it real.
Check out my new collaborated site: MNMLimpact.com
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
"Maybe I am missing something here but isn't it at least kind of cool that the rearrangement seemed to work?"
that's the beauty of chemistry you don't always have to understand why something works to know it works although it helps but some things are
difficult to explain.
for example isomerization of allybenzene with KOH.
they still don't have a solid mechanism for that.
you can learn a lot more by forging ahead despite your limited understanding.
biochemists on average only understand 5-10% of the system they are working with.
Give me librium or give me meth!
Patrick Henry....
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
The results are certainly encouraging, but the tricky part will be to remove unreacted aldehyde.
You could try oxidation with H2O2 or oxone, ideally following a published procedure. Maybe try it on neat aldehyde first, before destroying the
post-reaction mix. Unsurprisingly the resulting acid is called hydratropic acid. TLC control would be the best analytical method for this.
Other ideas would be derivation to other products, which are more easily separated. Maybe the bisulfite adduct forms more readily for the aldehyde and
it can be precipitated (and recuperated!) that way. But again, without some control via TLC, all is speculation.
BTW: The oil in your pictures looks very turbid. Did you not dry the organic phase, or is there some solid on the verge of precipitating? Maybe try to
focus the flask, not the background. 
|
|
|
Asphyxiated
Harmless
Posts: 20
Registered: 29-2-2012
Member Is Offline
Mood: No Mood
|
|
| Quote: |
BTW: The oil in your pictures looks very turbid. Did you not dry the organic phase, or is there some solid on the verge of precipitating? Maybe try to
focus the flask, not the background. |
Ill try to get some better pictures on later runs but the oil isn't turbid, its pretty much crystal clear, I did dry the organic phase, it just looks
like that in the pictures because, as you said, my camera kept focusing on the background and not the damn flask.
Also I am pretty sure that using the method that I did that the ketone has been separated from the aldehyde. At least thats what "Danilov and
Danilova, Chem. Ber. 60, 1050 (1927)" found when they did this procedure. Original paper in German here. Their end product was pure phenyl-2-propanone not a mixture of unreacted aldehyde.
From p2p I have seen before this looks like pure-ish p2p, not a mixture of unreacted aldehyde. As I found in my first run the H2SO4 does efficiently
oxidize the aldehyde to its carboxy acid, which should allow good separation via solvent extraction, I would think.
Anyway, more to come tomorrow (hopefully).
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
i don't think h2so4 is an oxidizing acid.
i would run tlc to be sure your not dealing with a mixture of products.
Give me librium or give me meth!
Patrick Henry....
|
|
|
Asphyxiated
Harmless
Posts: 20
Registered: 29-2-2012
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by jon | i don't think h2so4 is an oxidizing acid.
i would run tlc to be sure your not dealing with a mixture of products. |
Well hot conc sulfuric acid is certainly an oxidizing agent, but not at lower temperatures (source on that is wikipedia). However I am not sure how
'hot' is hot. The first time I ran this I definitely did not use enough ice in the second part of the rxn which lead to the solution reaching well
over 50 C which resulted in a bunch of acid crystals crashing from the aqueous phase and nothing to be found in the organic phase besides ~1ml of a
clear oil with a bp of 75C at reduced pressure (not sure exactly what my aspirator pulls).
But, I agree, I should run a TLC to be sure, since its not too difficult. I plan on running this reaction again tomorrow, will report back with the
results, hopefully some good pictures and perhaps some tlc results as well if I have time.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
No need for oxymoronic declarations like that. Things can not look pure-ish in chemistry unless you have analytical data. Analytical data is not
subordinate to wishful thinking.
| Quote: | | Maybe I am missing something here but isn't it at least kind of cool that the rearrangement seemed to work? |
No, it is nothing cool because we know that since 1927 and we still wait for you to contribute something of scientific value that would warrant the
presence of such a thread on this forum.
PS: Just one more attempt at pretending at being a drug cook and this thread goes to Detritus. I don't want to see any more drug cook terminology
here. Just one more "P2P" or anything like it and say by by to the thread. I already pointed you to the guidelines. I did so you would read them!
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Asphyxiated | | Well hot conc sulfuric acid is certainly an oxidizing agent, but not at lower temperatures (source on that is wikipedia). However I am not sure how
'hot' is hot. The first time I ran this I definitely did not use enough ice in the second part of the rxn which lead to the solution reaching well
over 50 C which resulted in a bunch of acid crystals crashing from the aqueous phase and nothing to be found in the organic phase besides ~1ml of a
clear oil with a bp of 75C at reduced pressure (not sure exactly what my aspirator pulls). |
I hope you realize that you are contradicting yourself?
On one hand you say that there is no aldehyde in the post-reaction mix, on the other hand you claim to have obtained "a bunch" of hydratropic acid
which can only come from hydratropic aldehyde?
What does the original literature say? I lack the motivation to struggle through it. I merely skimmed over it and it seems to say there are only
condensation products and phenylacetone, right? The phenylacetone was apparently identified by the refractive index (an annoying, but if done right
quite precise method - very temperature dependent) and the semicarbazone derivate.
Conc. sulfuric acid certainly is quite oxidizing at elevated temperatures - after all it dissolves the noble metal mercury and the semi-noble metal
copper. But once you reached the 50°C you had already dilute sulfuric acid. Moreover, I doubt that sulfuric acid is a clean oxidizer for organic
materials. It probably gives a mess. And you should have smelled SO2. So the nature of your white crystals is, so far, pure speculation as is the
purity of your distillate.
Yes, it can be tedious, but more analytics is needed!
|
|
|
Asphyxiated
Harmless
Posts: 20
Registered: 29-2-2012
Member Is Offline
Mood: No Mood
|
|
There is no contradiction, the first synthesis had nothing come over in the distillation of the organic phase besides the 1ml of clear oil but the
aqueous phase, upon cooling crashed out a ton of crystals. On the second synthesis attempt during distillation 10.5 grams (I now have weighed the
product, not just eyed the volume) of a bright yellow/green oil came over and I could only get a small amount of crystals to crash out of the aqueous
phase again.
I never claimed that there was no aldehyde in the post-reaction mixture but once it has been washed with DCM (or ether as the literature uses) then
there should be no aldehyde in the organic phase and 2-phenylpropionic acid in the aqueous phase, which must have been what crashed out in a great
quantity in the first synthesis attempt. I say it must have been 2-phenylpropionic acid and not 2-phenylpropionaldehyde because the aldehyde is
miscible with water and liquid at room temperature and since it is the only organic compound being used in this synthesis I can not think of anything
else that would have crashed out of the aqueous phase. I am aware that "can not think of" does not mean "was exactly this".
How have I contributed exactly zero to this synthesis? I have search long and hard for reports from people actually trying this out, besides the
original authors back in 1927, and found zero. Perhaps it was just never documented but well known to work to those "inside the circle" (whatever
circle that may be). I haven't claimed to be 100% sure that my yellow oil is 100% phenyl-2-propanone, I even agreed that I need to do TLC to be sure.
Concerning my statement of "it looks like fairly pure-ish phenyl-2-propanone I have seen before" I meant before it was so controlled and could just
acquire it from a chemical supplier. The bottle said "phenyl-2-propanone >= 96%", seems pretty pure to me. That old bottle, not my product.
Concerning the use of "p2p" instead of phenyl-2-propanone, so can I also not use MEK for methyl ethyl ketone? Or any other abbreviations for
substances that have already been identified earlier in the thread with their full chemical name? Or is it just phenyl-2-propanone that you want me to
spell out each and every time?
I did read the rules and I did not see anything about using abbreviations for compounds as a no no, I think I am being hen-pecked to death here. If I
am such a drag on this forum with my 3 threads then I guess just get rid of everything I put on the site. I thought it might be cool to have some kind
of 'first hand experience' log for this reaction which is almost completely undocumented except for its original paper for 1927.
| Quote: | | Just one more attempt at pretending at being a drug cook.... |
Whose pretending here? Do the pictures I posted show me "pretending" to do this synthesis? I don't understand what you meant by this at all. Just
because I don't plan on taking the next step of acquiring methylamine and making methamphetamine doesn't mean that I am "pretending" to do this. But
maybe thats not what you meant.
And finally I would like to say that I am whole heartedly sorry for any and all sleepless nights and migraines from the frustration that some people
seem to be having when reading my thread. Seriously, I am sorry, if you give me a po box ill mail you some Excedrin to help with that.
[Edited on 4/4/2012 by Asphyxiated]
|
|
|
Bot0nist
International Hazard
Posts: 1559
Registered: 15-2-2011
Location: Right behind you.
Member Is Offline
Mood: Streching my cotyledons.
|
|
Relax friend. I believe Nicodem was just trying to encourage an improvement in you scientific discourse. This isn't the chemistry I would do at home,
but I really appreciate you sharing your experimental data.
P.S. The hen-pecking may, in part, be do to your chosen subject of interest. Not the most popular "science experiments" 'round these parts.
U.T.F.S.E. and learn the joys of autodidacticism!
Don't judge each day only by the harvest you reap, but also by the seeds you sow.
|
|
|
aliced25
Hazard to Others
Posts: 262
Registered: 31-7-2010
Member Is Offline
Mood: No Mood
|
|
Oxidation of an aldehyde without touching a ketone, dunno... Perhaps Tollen's reagent? Possibly Nickel (II) oxide could be used to oxidise the aldehyde (it was suggested elsewhere to oxidise an aldehyde that was
prone to self-condensation), but that generally uses hypochlorite. Then again, that would presumably mean two acids if what you hope happened actually did happen, hydratropic and phenylacetic, which
goes some way to working out what is in the pot. If any chloroform is produced, obviously you must have a methyl ketone and the amount of chloroform would tell you what percentage of the mix of acids was presumably the source
of the chloroform.
I wonder if the acids would produce the alkene with one less carbon like hydrocinnamic acids (and a-alkylhydrocinnamic acids) are reported to do when
strongly heated? If they did, toluene and styrene could presumably be characterized by boiling point, oxidation of styrene to acetophenone and toluene
to benzoic acid, all sorts of wild fun. What is the boiling point of the various esters of the acids, can they be separated easily?
Look at old issues of J. Biol Chem, J. Chem Soc. or even Am. Chem. Soc., various authors did this sort of shit all the time. They rarely engaged in
bickering, name calling or snide remarks, nor did they get defensive and snarky when suggestions were made to them. There are other forums where
certain rules are not enforced, there are few forums with as many ACTUAL chemists as this one (I'm a member on other forums and I ain't a chemist, I
value the level of knowledge (and experience) on this board). If you wish to be a k3wl this isn't the right board for you, then again, if you wish to
actually conduct experiments, let me suggest you may be very, very lonely on other boards.
PS Presumably you are somewhere this is legal to do, if you aren't - taking photographs of yourself (and your hand) doing so - may not be very clever
or particularly healthy.
[Edited on 5-4-2012 by aliced25]
From a Knight of the Realm: "Animated movies are not just for kids, they're also for adults who do a lot of drugs." Sir Paul McCartney
|
|
|
Mildronate
Hazard to Others
Posts: 428
Registered: 12-9-2009
Member Is Offline
Mood: Ruido sintetico
|
|
Please somebody explain mechanism
Can anybody write/explain mechanism for Phenyl-2-Propanone by Rearrangement of 2-Phenylpropanal?
http://www.erowid.org/archive/rhodium/chemistry/phenylaceton...
|
|
|
woelen
Super Administrator
Posts: 7977
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Did you put any effort in this yourself? This post reeks of cookery and spoonfeeding and easily could end up in detritus.
|
|
|
Nicodem
|
Threads Merged
1-7-2013 at 06:11 |
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
What makes you think the mechanism has been studied? Have you done a literature search about it? I don't remember any such study. If all you want is
just some hypothetical reasonable mechanism (aka "arrow pushing"), then you can get this one (though I can think of related pathways). But
hypothetical mechanisms are not really good for anything except for building more hypotheses - at least until they experimentally prove themselves
useful.
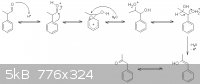
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
aliced25
Hazard to Others
Posts: 262
Registered: 31-7-2010
Member Is Offline
Mood: No Mood
|
|
Our friend Mildronate has a marked tendency toward requests for spoonfeeding. Not that others don't, but the majority of them at least put some effort
in. It is somewhat disturbing that the requests ALL seem to be inviting others to participate in the discussion of potentially illegal materials,
without stepping over the line to actual discussion of the same him/herself. Make of that what you will.
As to how to identify the product, might I take the opportunity to retract previous statements made as a result of having done some further research.
I would be very reticent to use the haloform reaction as it will give the benzoic acid, while oxidation with H2O2 will give the benzoic acid as well
(Jones, D.D., 1966 'The Alkaline Hydrogen Peroxide Oxidation of Phenyl-2-Propanones' PhD Dissertation, Georgia Institute of Technology (https://smartech.gatech.edu/handle/1853/5564)).
That doesn't mean the haloform reaction is necessarily a bad way to identify the product, benzoic acid could be isolated from the mix and it has a
boiling point that is different to hydratropic acid. It is just that I am not so sure that it will work effectively (both may give benzoic
acid).
[Edited on 1-7-2013 by aliced25]
From a Knight of the Realm: "Animated movies are not just for kids, they're also for adults who do a lot of drugs." Sir Paul McCartney
|
|
|









