| Pages:
1
2
3 |
spirit
Harmless

Posts: 8
Registered: 30-8-2012
Member Is Offline
Mood: No Mood
|
|
picric acid from aspirin
I need help. I have ran this reaction twice almost to completion. I have seen the color changes but have no precipitate.
I have extracted aspirin from 20, 325mg tablets. I have two procedures both describing the same process. Both are equally vague.
The first adds 5 grams aspirin to 80ml fuming h2so4 on water bath for fifteen minutes. Solution turns yellow. It is removed from heat and 15g KNO3 is
added in the additions with stirring. There's no brown gas evolved but there are fumes produced. After the additions solution turns red and with
stirring becomes yellow again. It is cooled to room temp and added to 1000ml h2o and crushed ice. Yellow crystals are supposed to precipitate and get
filtered. the yellow residue is washed twice with cold water to yield picric acid.
The second method starts by adding 5g aspirin to 80ml fuming h2so4 at 70deg c. Solution turns yellow. It is cooled to 50 def c. And KNO3 is added.
Solution turns red. Stirring until it returns to yellow. It is brought to room temp and added to 1000ml ice water. The ice is allowed to melt on it's
own and the yellow crystals that precipitate are filtered.
The first time there were no temps to follow and I believe I reduced the aspirin to acetic acid which is why there was no product. The second time
seemed to go fine but here I am after the ice melt with no precipitate and 1.5 liters of corrosives I don't want to filter if I've already bombed it.
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Use the search engine, there are probably more picric acid synths on this web site and any other compound, some very detailed indeed. Also during this
nitration both acetic acid and CO2 are given off so your comment "I believe I reduced the aspirin to acetic acid" probably indicates that you at least
hydroysed the aspirin to salicylic acid.
|
|
|
Mailinmypocket
International Hazard
Posts: 1351
Registered: 12-5-2011
Member Is Offline
Mood: No Mood
|
|
Why on earth are you using so much FUMING sulfuric? Where did you get your synthesis from?
*BTW this should be in the energetics section of the forum.
[Edited on 11-11-2012 by Mailinmypocket]
|
|
|
plante1999
International Hazard
Posts: 1936
Registered: 27-12-2010
Member Is Offline
Mood: Mad as a hatter
|
|
I already made 1 g of picric acid from aspirin. I didn't purified the aspirin. I didn't had any use for the picric acid so I just stored it under
water. I don't think I could use it for something, I just made the picric acid because I like how all old Lab had a bottle full of it, I also wanted
to make phenol from it but I found it to be quite difficult to do. 1g under water should be quite safe
I used 14 ml 96% H2SO4
2 g KNO3
1 g aspirin
First I dissolved the aspirin in the sulphuric acid and added the KNO3, it turned red. Then I heated it with an alcohol burner, It turned black and
made a lot of gas, after few minutes of heating the solution turned very light yellow. Then I added the solution on a 50ml ice cube and I waited until
it melted. I filtered the solution and I got 1 g of picric acid when dried.
WARNING!
Picric acid stain really strongly most organic material especially skin and nail! The yellow color fade take about 10 day for skin and doesn't for
nail until you cut them!
[Edited on 11-11-2012 by plante1999]
I never asked for this.
|
|
|
spirit
Harmless
Posts: 8
Registered: 30-8-2012
Member Is Offline
Mood: No Mood
|
|
Mailinmypocket- the first comes from an improvised munitions handbook. Most of the precursors are made from other sections in the book. I made my KNO3
only. It calls for boiling battery electrolyte for the fuming h2so4. I was attempting hardware grade that had been clarified by boiling with h2o2. I
think the volume it calls for is before boiling until fuming, all other procedures I've seen use around 80ml h2so4.
I noticed how saturated the solution became (that oily look) after the KNO3 addition. If it appeared saturated at this volume, maybe it wouldn't have
been able to dissolve a batch this size (roughly 5x plante1999 synthesis). off the top of my head I remember KNO3 dissolves around 1g/16ml h2o and I
used 15g...
good point about the energetics section, slipped my mind. my apologies.
Plante1999, thanks for sharing your experience. I will have to try your way. Were you able to see a precipitate in the solution before you filtered?
Of the techs i have one way insinuates a residue of picric acid, and the other crashes yellow crystals with the ice water. I'm just wondering if I
should be seeing 4-5g of material floating around before I decide to filter this beaker.
|
|
|
plante1999
International Hazard
Posts: 1936
Registered: 27-12-2010
Member Is Offline
Mood: Mad as a hatter
|
|
When I added the picric acid to the ice cube it change color from slightly yellow to a really bright yellow, after the ice cube as melted there was
yellow powder floating around. Filter while cold tough.
And please delete one of the two similar reply you made.
I never asked for this.
|
|
|
Mailinmypocket
International Hazard
Posts: 1351
Registered: 12-5-2011
Member Is Offline
Mood: No Mood
|
|
Battery grade sulfuric acid is 85% If not less, or around that. Boiling it until it fumes does not make it "fuming sulfuric acid" it just concentrates
it. Fuming sulfuric acid is made by adding sulfur trioxide to concentrated sulfuric acid (98%+) and having free SO3 in the acid. What you probably
have is pretty concentrated sulfuric acid, not fuming. Still, it seems to be a bit much as far as the synth is concerned.
[Edited on 11-11-2012 by Mailinmypocket]
|
|
|
spirit
Harmless
Posts: 8
Registered: 30-8-2012
Member Is Offline
Mood: No Mood
|
|
I realize it's garbage in garbage out. The improvised handbook has you boil until fumes because that might be all you can do in the field. I'm going
through the book just to see how realistic it's claims are and then I am trying it again for purity.
Might I have success dissolving the aspirin in h2o, generating SO3 FROM cupric sulfate, and bubbling with heat? Buying good acid isn't a problem, I
just want to make as much from scratch as I can first.
|
|
|
Nicodem
|
Thread Moved
11-11-2012 at 00:50 |
spirit
Harmless
Posts: 8
Registered: 30-8-2012
Member Is Offline
Mood: No Mood
|
|
I let the flask settle and there is a layer of black on the bottom which I think is the sulfuric acid contaminants. I can now see a bunch of particles
suspended in solution. Maybe I can decant some while leaving the black behind. Thanks for the help.
|
|
|
caterpillar
Hazard to Others
Posts: 472
Registered: 8-1-2012
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by plante1999  | I already made 1 g of picric acid from aspirin. I didn't purified the aspirin. I didn't had any use for the picric acid so I just stored it under
water. I don't think I could use it for something, I just made the picric acid because I like how all old Lab had a bottle full of it, I also wanted
to make phenol from it but I found it to be quite difficult to do. 1g under water should be quite safe
I used 14 ml 96% H2SO4
2 g KNO3
1 g aspirin
First I dissolved the aspirin in the sulphuric acid and added the KNO3, it turned red. Then I heated it with an alcohol burner, It turned black and
made a lot of gas, after few minutes of heating the solution turned very light yellow. Then I added the solution on a 50ml ice cube and I waited until
it melted. I filtered the solution and I got 1 g of picric acid when dried.
WARNING!
Picric acid stain really strongly most organic material especially skin and nail! The yellow color fade take about 10 day for skin and doesn't for
nail until you cut them!
[Edited on 11-11-2012 by plante1999] |
You had to take at least twice more KNO3. Check your numbers. And remember, that some excess of KNO3 (33% at least) is a must. My first attempt to
prepare picric acid was realized with the same mistake. Later I used phenol- 250 gr and 1000 gr of KNO3.
Women are more perilous sometimes, than any hi explosive.
|
|
|
Hawkguy
Hazard to Others
Posts: 326
Registered: 10-10-2014
Location: British Columbia (Canada eh!)
Member Is Offline
Mood: Body is Ready
|
|
I just did this, but the precipitate colour was that of white people, like a pinkish beige.... Could I have made a wierd isomer?
|
|
|
greenlight
National Hazard
Posts: 705
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
I tried a variation of the first procedure stated by Spirit a couple of years ago but had the same problem with no precipitate at all, just yellow
ice-water. I tried it two more times before after this before giving up, with more heating time while adding the ASA and slower addition time of the
Potassium nitrate with longer stirring and ended up with a small amount of picric acid but not worth the time.
I don't think there was enough Kno3 in the reaction but I will try again in the future with some users variations from the old E&W forum i have
written down, or if I can locate phenol.
[Edited on 8-11-2014 by greenlight]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Look for, "Rosco's Good Old Country Recipe for Picric Acid". It is basically foolproof as long as the instructions are followed. I used to try and get
creative with this synthesis back when my scientific background was such that most improvisations done by me were not improvements. Rosco's method
seems to be very nearly optimized.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
greenlight
National Hazard
Posts: 705
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Thanks, I will look for that document and try his synth if i can find it.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by Hennig Brand | | Look for, "Rosco's Good Old Country Recipe for Picric Acid". It is basically foolproof as long as the instructions are followed. I used to try and get
creative with this synthesis back when my scientific background was such that most improvisations done by me were not improvements. Rosco's method
seems to be very nearly optimized. |
I think this should help
http://www.sciencemadness.org/talk/viewthread.php?tid=389&am...
|
|
|
forgotpassword
Harmless
Posts: 47
Registered: 12-8-2014
Member Is Offline
Mood: No Mood
|
|
I was under the impression that the Aspirin had to be hydrolysed to Salicylic Acid before nitrating it
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
If aspirin is used instead of salicylic acid hydrolysis occurs in the hot sulfuric acid sulphonation stage of the process. The products of aspirin
hydrolysis are salicylic acid and acetic acid. The salicylic acid is then sulphonated to almost exclusively 5-sulphosalicylic acid from what I
understand.
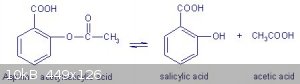 
During nitration the sulphonic acid group is easily substituted by a nitro group and finally the dinitro-salicylic acid is decarboxylated and the
final nitro group is added producing picric acid. The decarboxylation is witnessed as the foaming that occurs especially during the last third of the
nitrate salt addition.
[Edited on 9-11-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
TGT
Harmless
Posts: 46
Registered: 9-11-2014
Member Is Offline
Mood: No Mood
|
|
Picric acid is one of my favorite energetics to synthesize, second being ETN. I have made it many times over the last couple years and have now got
it down to perfection. I have used 70% nitric acid and also ammonium nitrate and have found no difference in yield or quality. One of the most
important attributes in a successful batch is temperature control vs time.
If anyone is interested in the details let me know. I have wanted to get it recorded for sometime as my notes are extremely messy, not to mention the
hole that goes through them from a large drop of sulphuric acid lol. If someone wants a write up it will push me to make a good copy. I
unfortunately have severe procrastination problems.
TGT
|
|
|
greenlight
National Hazard
Posts: 705
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
I would be very interested in a reading writeup of your picric acid synthesis from 70% nitric acid as i want to attempt to make this explosive in the
next week or two after I purchase some aspirin tablets to extract. I have tried it acouple of times before with low yields (almost nothing).
I will try yours since you say you have got it down to perfection if its uses acetylsalicylic acid.
|
|
|
TGT
Harmless
Posts: 46
Registered: 9-11-2014
Member Is Offline
Mood: No Mood
|
|
Okay, I will do a write up as soon as I can and post it. I need to record it anyways for my own personal notes so thanks.
TGT
|
|
|
TGT
Harmless
Posts: 46
Registered: 9-11-2014
Member Is Offline
Mood: No Mood
|
|
Well, here is half the procedure. I wrote it very detailed so that there would be hopefully no problems while performing this task. The next part
I'll post in a couple days. Also, I am sure there is many Picric Acid procedures here, but I just wanted to post the one I know for me to be the
best.
------------------------------------------------------------------------
Picric Acid from ASA Tablets / ASA Extraction
The following will describe in detail how to extract Acetylsalicylic Acid (ASA) from over the counter tablets and then finally convert it to Picric
Acid for use as a booster in high explosive caps. You can do this all in one run, but I like extracting the ASA to high purity first and then drying
for later use. You can use it before it dries, but for our purposes I will describe how to do it separately for clarity.
First you must find ASA tablets that contain the least amount of ingredients. The no name brands are usually best for this as they contain only Corn
Starch and Microcrystalline Cellulose as binders. Both binders are not soluble in alcohol and ASA is, so this will make extraction very easy. I will
be using a box with a count of 500 tablets each containing 325 mg. This will give us a theoretical yield of 162.5 grams of pure Acetylsalicylic Acid.
Materials for ASA Extraction:
• 2 1000 ml Beaker - Coffee grinder/Mortar and Pestle
• 2 Coffee Filters - Stir Stick
• Approximately 600 ml of Methanol - 600 ml cold Water
• 500 325 mg ASA Tablets - Ice or access to a fridge
Procedure:
- Crush ASA tablets into a fine powder using the Mortar and Pestle and/or a Coffee Bean Grinder.
- Place powdered tablets into a 1000 ml beaker and add 600 ml Methanol.
- Place ASA/Methanol mixture on a hot plate and heat until hot, but not boiling, while stirring well. Five minutes or so should be enough time to
dissolve the ASA. The ASA will dissolve, but the binders will not. Remove beaker from hot plate.
- Place a Coffee Filter on another 1000 ml empty beaker and filter the ASA/Methanol mixture and discard the binders and filter paper. Now there
should be only ASA and Methanol in the beaker.
- Place beaker back on hot plate and boil the liquid down to 150 ml. I use Methanol because it has a low boiling point and we want to keep the
temperature below 60 degrees Celsius so that the ASA does not hydrolyze and break down into Salicylic acid. For Picric Acid production it probably
doesn't matter, but for our purposes we will keep it as ASA. You can use Isopropyl Alcohol or Ethanol if you do not have Methanol, but still keep an
eye on the temperature and it will take longer to boil down. Make sure you do this outside or in a fume hood. The vapors are toxic and the liquid
flammable.
- Fill an empty 1000 ml beaker with 600 ml of ice cold water and crash the 150 ml of Methanol/ASA liquid into the water. You will instantly see
precipitate forming as ASA is not soluble in water. This also gives you very clean product instead of boiling to dryness. Also boiling to dryness
could cause the ASA to turn to Salicylic Acid.
- Stir gently and if still warm from the addition of Methanol place in a fridge for a few hours, then filter and dry. The crystals will be pure
white and fluffy. You can speed drying by dabbing and pressing the water out with a dry paper towel. Usually the crystals will not stick to the
paper. You will have very pure Acetylsalicylic Acid to use for our next step. I like to do this procedure on day 1, and then on day 2 start with the
Picric Acid.
to be continued......
|
|
|
greenlight
National Hazard
Posts: 705
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Thanks I will do this step now and as soon as you write up the second part I will give it a go and post results to see if I can get the same high
yields as you.
I like the idea of methanol instead of isopropanol to extract the ASA because it is much cheaper here to buy than the isopropyl alcohol.
|
|
|
TGT
Harmless
Posts: 46
Registered: 9-11-2014
Member Is Offline
Mood: No Mood
|
|
Picric Acid from ASA Tablets / Part 2
The following will describe in detail how to convert the Acetylsalicylic Acid (ASA) from the previous procedure to Picric Acid, a very powerful
explosive material. This is a very dangerous synthesis and should be performed by someone with adequate experience in energetic materials. It should
also be performed in a fume hood or outside, never indoors without proper ventilation. There is always a chance of a runaway reaction when adding the
Ammonium Nitrate or Nitric Acid (depending on which you choose to use). This happens when the temperature is not kept in the proper range or the
materials are added too quickly. That is why it is always a good idea to have a large bowl of water/ice cubes with sodium bicarbonate mixed in as a
safety precaution. If something unexpected should happen you can always dump the project into the bowl and avoid being injured.
In the previous experiment we purified more ASA than is needed as the following procedure will only use 35 grams of ASA. The reason for this is to
keep the experiment relatively small and simple. After experience in synthesizing Picric Acid you can scale up the procedure. Scaling up is not
mathematically consistent and required experimentation. That is why at the end of this document I added some values to follow for larger batches that
I have previously completed with excellent yields.
One last important note before we start. In this procedure you can substitute Ammonium Nitrate for 70% Nitric Acid. I have found that 1 milliliter
of 70% Nitric Acid is equal to 1 gram of pure Ammonium Nitrate. For every gram of ASA you will need 2 grams or 2 ml of 70% Nitric Acid. Sulfuric
Acid will vary with use of Ammonium Nitrate, Nitric Acid or batch size. For this experiment we will be using Nitric Acid.
Materials for Picric Acid Synthesis:
• 250 ml Round bottomed flask - 500 ml Graduated Cylinder
• Thermometer - Glass Stir Stick
• Hot Plate - Hot Water Bath
• 35 grams of ASA - 70 ml of 70% Nitric Acid
• 95 ml of 96% Sulfuric Acid - Digital Scale
• 2, 1000 ml Beakers - Filter Paper
Note: If using Ammonium Nitrate in place of Nitric acid, add 110 ml of sulfuric acid. The rest of the measurements are to stay the same. Just make
sure with every addition of Ammonium Nitrate you mix extremely well and dissolve it all before adding more.
Procedure:
- Set up the hot water bath and place on hot plate. Preheat the water to 70 degrees Celsius so that it stays at this temperature consistently. I
personally didn’t use a hot water bath. Instead I used a coffee mug heater that maintained my temperature in the proper range and I found this
worked well. To be safe use a water bath, but use your own judgment.
- Add 95 ml of Sulfuric Acid to the 250 ml round bottomed flask and place it in the hot water bath.
- Weigh out 35 grams of ASA and set aside.
- With your glass thermometer measure the temperature of the Sulfuric Acid and when it reaches 65 to 70 degrees Celsius start the addition of
powdered ASA. Add around a gram at a time while stirring or swirling and watch your temperature. The temperature will increase slightly with each
addition, but not nearly as much as when the Nitric Acid is added. Make sure that all of the ASA is dissolved well into the Sulfuric Acid before
adding more. Consistent stirring or swirling is very important to avoid hot spots and to dissolve each addition properly. Do not let your
temperature increase beyond 95 degrees Celsius. You should try to reach the 95 degree mark at the end of ASA addition.
- After the ASA addition has completed continue heating at 95 degrees Celsius for 45 minutes while stirring or swirling occasionally. The additional
heating will sulfonate and decarboxylize the ASA. If your acid is clean you should see a color change from red to very dark brown or black. You
probably will not see this color change if your Sulfuric Acid is hardware store grade. Just follow the temperatures and times listed and it will be
fine.
- After 45 minutes of heating remove the solution from the hot plate and let it cool down to around 30 to 35 degrees Celsius. After it has cooled to
this temperature it is time to add the Nitric Acid.
Note: If your using Nitric Acid over 75% pure you might want to start the addition at room temperature or cooler. The higher the percent the lower
your starting temperature should be. The beginning temperature is for 70% because that is what I used for this synthesis.
- Measure out 70 ml of Nitric Acid using your graduated cylinder.
- Add extremely slow your Nitric Acid a few ml at a time to your Sulfuric Acid/ASA solution while stirring or swirling continuously. At the start do
the addition of Nitric Acid without the hot water bath and keep an eye on the temperature. Try to increase the temperature gradually so that the
beginning nitration is started at 35 degrees while the end of nitration reaches 85 to 90. Use the hot water bath if necessary. This temperature
range works reliably well and is much safer than heating too hot. Keep in mind there will be some red Nitrogen Dioxide gas with each addition, but if
done slowly this should be kept to a minimum. Near the end of the Nitric Acid addition the bubbling should slow and the reaction mixture takes on a
canary yellow to tomato sauce red color, depending on batch size. The entire Nitric Acid addition should take around half to one hour to complete,
but this can vary. Remember stirring or swirling is essential.
- Now that the Nitric Acid addition is complete, set up the hot water bath to a consistent temperature of 95 degree’s Celsius and heat the reaction
mixture/beaker while swirling often. Continue heating for no less than 1 hour at this temperature. This will complete the reaction. Make sure you
always keep a good eye on what is happening and never leave the chemicals unattended. Also, be sure to have your safely bowl of water and Sodium
Bicarbonate close by as described earlier. This was not added to the materials list, but should be considered important.
- Now let the reaction mixture cool to room temperature. You will start to see the mixture thicken up and crystals will begin to precipitate.
- Fill a 1000 ml beaker with 500 ml of ice cold water. Next, pour the reaction mixture into it. It will turn a beautiful shade of canary yellow.
Picric Acid will begin to drop out of solution. It is very important to leave this beaker in a cool place for 24 hours. A refrigerator would be
ideal. Picric Acid crystals sometimes take a long time to form and the longer you wait, the better the yield.
- After the 24 hours filter your crystals with filter paper and the other empty 1000 ml beaker and now you have crude Picric Acid. Wear gloves and
do not spill or drip any as it is extremely hard to remove, especially from skin. My lab bench is now a permanent shade of yellow to remind me of my
clumsiness. When finished you can press dry the crystals with a paper towel and place in a warm dry environment. Picric Acid is not extremely
hydroscopic so it should dry to a nice powdery consistency after crushing. Even at this stage it is relatively pure and can be used for most
purposes. In part 3 we will be recrystallizing them to get pure crystals of Picric Acid that can be safely stored for many years.
To be continues…
------------------------------------------------------------------------
Sorry this second part took so long. It was late when I finished it, so if anyone see's any mistakes please let me know. I tried to write it as
accurate as I could from my messy notes. I have tried this numerous times and is always reproducible. Any questions let me know.
TGT
[Edited on 22-11-2014 by TGT]
|
|
|
greenlight
National Hazard
Posts: 705
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
I just did the second step and everything went perfect but I just have a question about the last part of the nitration.
I added the 70% Nitric acid from an addition funnel in small portions over a 40 minute period and completed the addition fine. After the HNO3
addition, I placed the reaction mix which was 80. C on a heat bath to bring it to 95.C for the hour. When it got close to 95.C the reaction mixture
started to give off continuous small amounts of fumes of red Nitrogen dioxide but I continued anyway. Is this normal?
After it had been on the heat bath for about 15 minutes it started giving off more Nitrogen dioxide and went from tomato red to orange with small
crystals of picric acid coming out in the mixture.
I took it off heat when this started happening and more crystals came out until it all turned into a slurry of crystals. I dumped it into ice water
and it is still precipitating.
The synthesis worked so its not a problem I was just wondering if it has happened to you before?
I was thinking maybe I didn't need the extra nitration time because I'm using reagent grade acids, or maybe my stir bar had small particles of metal
it had picked up off the benchtop on it and they reacted with the Nitric acid?
|
|
|
Bert
Super Administrator
Posts: 2821
Registered: 12-3-2004
Member Is Offline
Mood: " I think we are all going to die. I think that love is an illusion. We are flawed, my darling".
|
|
| Quote: |
the reaction mixture started to give off continuous small amounts of fumes of red Nitrogen dioxide but I continued anyway. Is this normal?
|
This reaction reliably produces nitrogen oxides, hence the frequent mention of needing a proper fume hood or outdoor setup/fan for vapor extraction
and protective gear (mask with correct filters for NOx and sufficient capacity to carry you through the exposure time) in order to avoid injuring or
killing yourself.
You DID provide for fume removal and wore protective gear, correct?!
If not, PLEASE tell someone responsible what you just did, and be aware that pulmonary edema from such vapors often takes a while after exposure to
become obvious! We don't need any MORE dead amateur chemists.
| Quote: |
Acute Exposure Nitrogen dioxide is thought to damage lungs in three ways: (1) it is converted to nitric and nitrous acids in the distal airways, which
directly damages certain structural and functional lung cells; (2) it initiates free radical generation, which results in protein oxidation, lipid
peroxidation, and cell membrane damage; and (3) it reduces resistance to infection by altering macrophage and immune function. There may be an
immediate response to exposure to nitrogen oxide vapors that may include coughing, fatigue, nausea, choking, headache, abdominal pain, and difficulty
breathing. A symptom-free period of 3 to 30 hours may then be followed by the onset of pulmonary edema with anxiety, mental confusion, lethargy, and
loss of consciousness. If survived, this episode may be followed by bronchiolitis obliterans (fibrous obstruction of the bronchioles) several weeks
later. Any of these phases can be fatal.
|
RIP myfanwy
[Edited on 23-11-2014 by Bert]
Rapopart’s Rules for critical commentary:
1. Attempt to re-express your target’s position so clearly, vividly and fairly that your target says: “Thanks, I wish I’d thought of putting it
that way.”
2. List any points of agreement (especially if they are not matters of general or widespread agreement).
3. Mention anything you have learned from your target.
4. Only then are you permitted to say so much as a word of rebuttal or criticism.
Anatol Rapoport was a Russian-born American mathematical psychologist (1911-2007).
|
|
|
| Pages:
1
2
3 |









