| Pages:
1
2
3 |
gravityzero
Hazard to Self

Posts: 79
Registered: 14-6-2013
Member Is Offline
Mood: forgetful
|
|
After reading the thread in full, it is still unclear if anyone is able to get to Styrene Oxide simply using a peracid, such as performic acid or
peracetic acid.
It is assumed it would work, but OrgSyn uses perbenzoic acid which I had never heard of until today. Not really a miracle or anything.
If anyone can confirm a success using a more convenient peracid or oxone that would be helpful.
Then it is stated using silica gel to isomerize the oxirane to the aldehyde.
Sorry for any spoonfeeding. Just want to make sure I'm understanding all this.
|
|
|
NitreRat
Harmless
Posts: 45
Registered: 22-1-2015
Location: Cyberspace
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by gravityzero  | | After reading the thread in full, it is still unclear if anyone is able to get to Styrene Oxide simply using a peracid, such as performic acid or
peracetic acid. |
I have performed a few epoxidations of alkenes with peracids (including in-situ peracetic acid), albeit in a formal lab setting. Peracids are strong
oxidising agents so the actually reaction occurs quite readily, the real difficulty is trying to isolate the epoxide before any nasty nucleophiles can
attack the poor epoxide and break open its ring with their electrons...
Because of the inherent acid present with peracid epoxidations, any water, amines or alcohols present in the reaction mixture can react with the
epoxide after it's formed - creating alcohols and vicinal diols. These alcohols and diols/glycols can then be oxidized and cleaved by the peracid
forming aldehydes, ketones, hydroxy-ketones, diketones and carboxylic acids.
If you're using anhydrous peracetic, performic or (m-chloro)perbenzoic acid the hydrolysis can be avoided. If you're doing a one-pot in-situ peracid
epoxidation with H2O2(aq) + acid + alkene, you will inevitably end up with the hydrolysis and oxidation products. With styrene,
I would expect phenylacetic acid, phenylglyoxylic acid and benzoic acid to be the main products of this reacation.
[Edited on 11/3/2016 by NitreRat]
|
|
|
purplephanta
Harmless
Posts: 1
Registered: 26-6-2015
Member Is Offline
Mood: No Mood
|
|
Hello Phendrol, or anyone else for that matter,
Have you had success using the method referenced by Nicodem and TheCatalyst?
DO.108/00397919508011817 (http://dx.doi.org/10.1080/00397919508011817)
If so, what were the pitfalls;
Is the reaction not scalable?
Is very pure, anhydrous styrene oxide required?
Can the ethyl acetate be recycled or is it consumed?
Thanks
|
|
|
madcedar
Hazard to Others
Posts: 116
Registered: 10-9-2009
Member Is Offline
Mood: No Mood
|
|
styrene oxide to phenylacetaldehyde via the pinacol rearrangement?
In theory, mixing styrene oxide and a non-halo acid (H2SO4) should give styrene glycol. A halo acid such as HCl will give a
halohydrin instead.

On further mixing and heating phenylactaldehyde should be the product (see this Youtube video on the Pinacol Rearrangement).
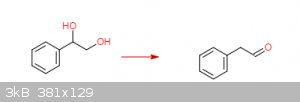
I'm assuming roughly following this procedure in the attached pdf (Organic Syntheses, Coll. Vol. 1, p.462 (1941); Vol. 5, p.91 (1925)) and
substituting an equal molar amount of styrene oxide for pinacol will give the desired results.
I don't have any glassware to try this out myself but any comments on the theory are welcome.
Attachment: Pinacol Rearrangement cv1p0462.pdf (298kB)
This file has been downloaded 677 times
[Edited on 18-6-2017 by madcedar]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
I ran across this paper a while back. I can't read most of it, but according to the authors, it's possible to do the rearrangement from styrene oxide
to phenylacetaldehyde using a catalyst consisting of nitric acid on activated carbon in ethyl acetate at 75 C:
Preparation of Phenylacetaldehyde and 1, 3-Dioxolanes from Styrene Oxide with Mineral Acid-Treated Activated Carbon Catalyst
|
|
|
Chemi Pharma
Hazard to Others
Posts: 349
Registered: 5-5-2016
Location: Latin America
Member Is Offline
Mood: Quarantined
|
|
Better produce phenylacetaldehyde from the isomerization of styrene oxide with silica gel in acetone, at room time for an hour, like said in the paper
i bring below.
I think the better route to phenylacetaldehyde is dissolving styrofoam in acetone, mix with MgSO4 as a cataliser and distill at 250-300ªC the styrene
monomer with the acetone all onto the same flask.
The styrene in acetone obtained react with TCCA to form the chloridrine and further with KOH to form the epoxide.
Finally, proceed with the epoxide isomerization with silica gel as I said above.
Here's the papers:
Attachment: styrene oxide to phenyl acetaldehyde with silica gel.pdf (981kB)
This file has been downloaded 889 times
Attachment: styrene from polystyrene.pdf (1MB)
This file has been downloaded 634 times
Attachment: styrene to styrene oxide with TCCA and KOH.pdf (234kB)
This file has been downloaded 815 times
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
I've read a number of interesting papers about isomerization of styrene oxide with exotic catalysts, and there is a U.S. patent regarding the use of
silica gel in the vapor phase, but doing it with silica gel in half an hour at room temperature... is that really possible?
Apparently, it is pretty easy to isomerize styrene oxide to phenylacetaldehyde in a bomb reactor with heat:
US3860614A - Thermolysis of Styrene Oxide
Attachment: Thermal Rearrangement of 1,2-Epoxyethylbenzene.pdf (252kB)
This file has been downloaded 500 times
I'm not terribly familiar with sealed tube reaction protocols, but I think the thermolysis could be performed using the ol' toaster oven with a long
extension cord trick and some sandbags. I'm not seeing why you couldn't use an unlined iron pipe sealed with plumber's tape to contain the reaction,
although I would advise extreme caution of course.
If you wanted to get really fancy, you could use an autoclave or a Parr reactor.
|
|
|
notoxicshit
Harmless
Posts: 37
Registered: 18-4-2016
Member Is Offline
Mood: No Mood
|
|
Works in acetone over silica gel in high yield. Forgot the name of the paper though it should be easy to find.
However they dont mention how dry the acetone has to be. That may be the reason I failed once but I got too confident and didnt check before the
follow-up reaction.
But it seemed legit.
Report back please.
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
Chemi Pharma posted the paper directly above. Silica gel 60 is pretty expensive... I'd be more inclined to try heat/pressure... the temperatures
required are nothing ridiculous, and no fancy apparatus is required.
Nicodem mentioned an expired Dow Chemical patent on a continuous vapor phase process for isomerizing styrene oxide to phenylacetaldehyde. It uses silica gel and water. The process looks a little
involved, but it might be useful if you want to produce more than a liter or so of phenylacetaldehyde.
|
|
|
Fyndium
International Hazard
Posts: 1192
Registered: 12-7-2020
Location: Not in USA
Member Is Offline
|
|
I have been on this project since last weekend and I have some issues.
I prepared the styrene oxide by TCCA method, but I did not vacuum distill it, instead I just removed acetone, washed it with brine and dried on
calcium sulfate.
Next I attempted the rearrangement by using 1:1 per weight of silica gel 60 (for chromatography) and the prepared oxide, but instead of the article I
accidentally used only less than half of the acetone. I stirred the mixture for half an hour and then vacuum filtered the silica gel off. I then
removed the acetone and tried to make bisulfite adduct for the aldehyde, but I have zero reaction.
Thing I did not check was pH after brine washing and drying. I wonder if residual NaOH from TCCA reaction could hinder the silica gel acidity?
[Edited on 23-7-2020 by Fyndium]
|
|
|
Fyndium
International Hazard
Posts: 1192
Registered: 12-7-2020
Location: Not in USA
Member Is Offline
|
|
Second attempt. I vacuum distilled the styrene oxide succesfully and attempted a rearrangement. pH was in control, but zero reaction with bisulfite
adduct was detected. Possible issues could be that acetone was not dry enough and it could have prior contaminated the silica gel as well, and second
cause that could have actually destroyed the aldehyde was distilling off the acetone. I think I should give it another try and use carefully dried
acetone and dry the silica gel prior, and instead of distilling acetone, crash the aldehyde out with water and reclaim the acetone later.
Edit: now when I fried silica gel in oven, my entire house smells of phenylacetaldehyde. So there sure is some of it, but maybe the reaction time was
too short or something stopped the reaction, because according to the article the isomerization should take only 30 minutes.
[Edited on 25-7-2020 by Fyndium]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
How do you know that you have successfully synthesized the epoxide? For that matter, how do you know that you synthesized the halohydrin? I have had
my eye on this synthesis for a while, but I planned to use DCCA or bromine water instead of TCCA because I never seem to have much luck with it.
I probably wouldn't use acetone as the solvent. Chloroacetone is quite nasty, and bromoacetone has been used as a war gas. I don't think TCCA reacts
very quickly with acetone at neutral or basic pH, but even small amounts of halogenated acetone are to be avoided. Also, the haloform reaction would
be a side reaction that would decrease the pH and consume the halogen.
|
|
|
Fyndium
International Hazard
Posts: 1192
Registered: 12-7-2020
Location: Not in USA
Member Is Offline
|
|
I have been under impression that the synthesis described in a link at the first post of this topic is valid. I have done it multiple times with
results matching the description, and density and bp of styrene oxide seems to be where it should be. I just finished vacuum distilling this presumed
epoxide and I came here to check what pressure my vac pump is able to generate and at 104C it seems to be 70mbar. All the leaking joints seem to be
taking their toll.
Of course there is a possibility that the synthesis is bogus. It wouldn't be the first time. Also the silica gel step sounds fishy because it just
seems too easy. I've got used that in chemistry to do a simple synthesis you will need five of the most watched reagents that are also contact
poisons, will detonate upon their own crystal weight and are hypergolic, and probably sublime away if you don't use them immediately.
Anyways, I'll get back on the isomerization tomorrow. I will make sure all the reagents and equipment are dry, and I was planning on taking samples at
½h, 1h, 2h, 3h, 6h and 12h to see if anything happens. The way to test aldehyde is with bisulfite adduct. What I've read elsewhere, it should react
readily upon mixing if pH is neutral. So far I haven't got any reaction whatsoever.
[Edited on 25-7-2020 by Fyndium]
|
|
|
Fyndium
International Hazard
Posts: 1192
Registered: 12-7-2020
Location: Not in USA
Member Is Offline
|
|
So far nothing. 30 and 60min samples taken, filtered, washed with water to crash the aromatic out of acetone and applied to bisulfite solution.
EDIT: I made a control with acetone and also added ethanol. It seemed to be heating a little, but nothing formed. I read elsewhere that someone had an
issue with potassium metabisulfite, which I've got.
EDIT2: Now I got it working. It needed some alcohol.
[Edited on 26-7-2020 by Fyndium]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
Cool. This is actually second on my list of syntheses to try when I get a new lab space. I believe you are the first person on this forum to get it to
work. It's worthy of a writeup in Prepublication.
|
|
|
Fyndium
International Hazard
Posts: 1192
Registered: 12-7-2020
Location: Not in USA
Member Is Offline
|
|
I might give it a shot when I perfect the process, like ratios, timing, etc and see how the adduct is released and I also want to be totally sure the
thing is real, because once I got it working it seems rather easy and fun part seems to be that a batch I made earlier and - luckily - left undisposed
also precipitated white solids when adducted, so water content or purity of the oxirane does not seem to be a critical matter, although it could
probably lower yields a lot. I had to basify it once during a batch because I used too much liquid and the adduct just went sort of suspension, so I
decided to cut it because bicarb is a lot cheaper and abudant than sulfite and added a good amount of sat bicarb and oil accumulated to the top.
I left a test batch sit in a jar overnight and it had a bulk of crystals hanging from the organic layer which sunk down upon disturbing the jar a
little. I suppose the adduct should be given time to form in this instance, or give it a good stir, like an overhead stirrer to agitate the whole
solution. On the other hand, the crystals seem to be the larger the slower they form, hence easier to separate and purify. Dumping a fast adduction
will form a suspension that just slips through filters, but it seem to settle eventually when left undisturbed and coagulates so that it can be
decaned and filtered. When I made cyanides a long time ago I lost probably almost half because I did not give it enough time to coagulate so patience,
or donothingness seems to be much more beneficial than pushing it. I think when finished the organic layer could be pipetted or suctioned off and then
the liquid decanted and the adduct dumped and vacuum filtered and washed with ethanol and or ether. The adduct was pale yellowish when filtered, but
turned pure white when rinsed with ethanol, and it was left to dry.
Downside is the smell. It's not bad, it's actually very nice - at first. But then it gets so strong and persistent it just annoys the hell out of you
- and the best part is you can still sort of smell or sense it even when you have left the shop for hours. I can still smell it in my fingers and
every time I touch my face it's just sweet flowers lol. Rinsing and cleaning all the ware and managing the waste liquid that has touched it will be a
mess of it's own. Dump it down the drain? No, it will reek through the piping and interceptor and you and probably your neighbors can also enjoy the
sweet smell.
But in the end, the smell of flowers was a smell of victory for me the moment I smelt it first.
[Edited on 27-7-2020 by Fyndium]
|
|
|
Fyndium
International Hazard
Posts: 1192
Registered: 12-7-2020
Location: Not in USA
Member Is Offline
|
|
I wonder what went wrong.
I attempted an adduct purification of the aldehyde and prepared it according to the Benzaldehyde from toluene with UV catalyst prepublication.
- I first prepared a potassium metabisulfite solution, added the organic layer and added ethanol until no more precipitated. I made a control without
organic layer and the sulfite did not precipitate. I vac filtered the white mass and washed it with more ethanol and dried it.
- I then prepared a sodium carbonate solution according to the same topic and I dissolved the adduct into it. It fizzled as it dissolved, and a very
little oil precipitated, the solution remaining cloudy and yellow. I attempted to extract it with ether, but upon concentrating it I obtained some
sharp smelling liquid that was nowhere the smell of aldehyde.
Now I wonder if I ever made phenylacetaldehyde in the first place? The smell is there, though. Is the adduct possibly made from something else? I
can't figure out what else could react with the bisulfite, the ethanol I used is leftovers from my own production so even MEK is out of question and
the amount of adduct formed is clearly much more than anything what could be the result of an impurity. I distilled and washed the organic layer with
brine multiple times to extract any remaining water and acetone prior to adduction. Or is there something wrong with the breaking of adduct? I tried
adding more carbonate and even hydroxide but nothing happened.
The breaking of adduct should be clear as a day, but nope, I guess. For the record, the adduct had strong odor of SO2 at least when it was still
moist, similar to bisulfite solution.
I would really appreciate a help with this, I've putten some effort into it and made it this far.
|
|
|
clearly_not_atara
International Hazard
Posts: 2691
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Does it smell burnt, stinging, smoky? Could be an aldol product, they often smell that way.
Unfortunately this is a particularly reactive aldehyde! One way to confirm and stabilize is to convert directly to the nitrile with hydroxylamine and
tungsten-tin hydroxide (easily prepared; see ref att.)
Attachment: yamaguchi2007.pdf (108kB)
This file has been downloaded 367 times
[Edited on 04-20-1969 by clearly_not_atara]
|
|
|
Fyndium
International Hazard
Posts: 1192
Registered: 12-7-2020
Location: Not in USA
Member Is Offline
|
|
Not at all burnt, just sulfur dioxide smell. You know it when it burns your throat and irritates your eyes, you cannot mistake it. Same smell as from
solution of sulfite. Only other smell it carries is sweetish, aldehyde-like smell, a very faint one.
Would it be possible that the aldehyde suffers from boiling off acetone? I would prefer to skip the adduct phase if possible, it is arduous and
costly, and if the isomerization step itself works above 92% yield, it should be pretty much useful as is, considering it will carry polymerization
products as impurities due to reactivity anyways. Distilling is of no use though, since the major impurity would be styrene oxide anyways, which has
the exact same boiling point and density. I have noted, by the way, that the aldehyde appears to be oxidising into what is probably phenylacetic acid,
since it obtains a different, but still strong odor profile when it is exposed to atmosphere, eg. when droplets of solution is splashed on surfaces,
etc.
If I just had hydroxylamine at hand. Are there any other methods to determine or purify the aldehyde from styrene oxide? Due to the isomer form, they
are practically the same by their physical properties, only apparent difference is the reactivity towards adduct. The styrene oxide itself appears to
be a volatile compound on it's own, it is stated that even trace amount of acid with water will turn it into phenethyl glycol, and if hydrolysis is
not possible, it will isomerize into aldehyde. I presume, that trace amount of water will cause hydrolysis until it consumes all the water and after
that only isomerization will occur?
Due to factors, I have vacuum distilled the oxide prior to isomerizing to neutralise it from the sodium hydroxide treatment and other trace
impurities. I use calcium sulfate to dry acetone and the oxide, since magnesium sulfate is slightly acidic.
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
In air? Try making the oxide or aldehyde a different way and compare?
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
I would probably suggest the bisulfite adduct-forming procedure written by Eleusis as the easiest and highest yielding, but I would suggest using
methanol instead of ethanol since methanol is cheaper and less likely to contain problematic denaturants.
There was some discussion on here recently about quantitative determination of carbonyls, and a reagent which can be used for that purpose is
2,4-dinitrophenylhydrazine. It's not the easiest thing to find, but it's not unaccessible in the U.S.
[Edited on 30-8-2020 by JJay]
|
|
|
Fyndium
International Hazard
Posts: 1192
Registered: 12-7-2020
Location: Not in USA
Member Is Offline
|
|
It seems that in every patent concerns the proneness to polymerization both the oxide and the aldehyde. Hence the aldehyde is prepared in solutions
containing 0.1mol concentrations and the silica gel method also requires 20:1 ratio of acetone. Theis could indicate that aldehyde was formed, but
proneness to polymerization and aldol could cause major part to turn into other stuff. Also catalyst deactivation by polymers could be a factor, I
suppose it could be purified by soaking in pure acetone.
https://doi.org/10.1016/j.apcata.2010.06.005
I have tried both, the patent amount and more concentrated solution and both indicate by smell, but no separate tests were made though, they both
reated with adduct.
Over-oxidation to acid could also be a factor.
It still bothers me though how such a large quantity of mass precipitated if it was not adduct, and basically nothing resulted when it was dissolved
with Na2CO3?
The next test I will try:
- soak and wash the silica gel in acetone and dry it at 120C for 2 hours prior to use
- dry acetone with magnesium sulfate and calcium sulfate and redistill
- when finished and filtered off the acetone, I have two choices:
1) crash out the product by adding water until phase separates
2) distill off acetone
Which one should be better? I would actually opt for 1, because it can be done in room temp and it is basically instant, while distilling exposes the
product to a very long duration of heat and it concentrates it as such up to a pure product. It would need to be washed anyways in order to remove
rest of the acetone, unless it were to be extracted by vacuum.
1 liter of acetone would hold 50 grams of styrene oxide and 50 grams of silica gel. As such, I would need to measure the amount of water needed to
crash out the product, but I've done it twice and it was successful.
I was also wondering if the reaction could be just carried out in a chromatography tube packed with silica, as the article originated from. The
concentration of silica apparently is trivial, but the ratio of oxirane/aldehyde to solvent seems critical to prevent any side reactions. This way,
even larger amounts of liquid could be handled with small amount of silica gel, and being protected from air and moisture as well. More testing, I
presume..
[Edited on 30-8-2020 by Fyndium]
|
|
|
clearly_not_atara
International Hazard
Posts: 2691
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Unfortunately this never worked out. The product is pretty unstable, and the reaction conditions for the pinacol rearrangement are roughly identical
to the ones for the undesirable aldol condensation. In particular, the pinacol intermediate (the carbocation) is identical to the intermediate in
acidic enolization.
Suppose that instead the pinacol rearrangement is carried out in ethylene glycol (itself resistant to the reaction) as solvent and hopefully most of
the phenylacetaldehyde were trapped as the ethylene acetal in situ. Then you just need a mild acetal deprotection.
There's an Indian paper claiming deprotection by chloral hydrate in hexane, but it sounds a little too good to be true.
[Edited on 04-20-1969 by clearly_not_atara]
|
|
|
| Pages:
1
2
3 |









