mayko
International Hazard

Posts: 1218
Registered: 17-1-2013
Location: Carrboro, NC
Member Is Offline
Mood: anomalous (Euclid class)
|
|
L-systems for algorithmic molecule generation
thought I would share an idea I'd been kicking around, finally got around to putting some code together...
______________________________
Abstract
An algorithmic chemistry is presented based upon Lindenmeyer systems. A family of organic molecules, including noncyclic alkanes, n-ols, polyethers,
and polysulfides, is generated by a simple formal system. The system treats molecules as networks with a tree topology; these can be easily generated
by a string-rewriting system.
Introduction
Artificial chemistry is the application of modern computational techniques, especially those influenced by complexity and artificial life, to study
abstract chemistries in silico. Examples range from models of molecules as networked finite state machines [1, 2] to graph-based quantum models. [3]
Lindenmeyer systems, or L-systems, are algorithmic descriptions of plant structure; because plants are topologically tree-like, they can be described
as a string rewriting system [4] An L-system consists of an alphabet of symbols V, an axiom string ω, and a set of production rules P. The
production rules are applied iteratively, to generate a series of new strings ω, P(ω),P(P(ω))....
Consider V = {a,b}, ω = a, and P={a -> ab; b->a }
This generates a sequence of strings a, ab, aba, abaab, abaababa..... As it happens, this provides a developmental model for filamentous algae. [4]
More complex structures with branching can be generated by including in V symbols for push (here, ‘[‘) and pop (‘]’). The above example
includes a set of deterministic production rules, but it is fairly straightforward to make a set of probabilistic production rules and thus create
stochastic output.
In the current implementation, molecules are generated by L-systems, thus defining a set S of well-formed strings; S = {Pn(ω): n=0,
1....} The starting axiom ω is taken to be a methane molecule, establishing a trivial saturated carbon backbone. The set of production rules
includes a chain-growth rule, whereby a hydrogen atom is probabilistically replaced with a methyl group, and a rule to generate alcohols by replacing
hydrogen atoms with hydroxyl groups. The nature of the production rules imposes a treelike topology on the set of generated molecules; ie, there are
no cyclic molecules, and they may be nontrivial to implement within the current programming scheme. This is a formal model; it is not intended to
mimic actual reactions but to explore the space S. Moreover, a molecule which is well-formed as a formal string need not be a physical molecule; the
production rules used here allow for arbitrarily long polyperoxides R-O-O....-R, which are not likely to be stable in reality. The current
implementation lacks more advanced functional groups such as unsaturated carbons, halogens, etc.
A Google/Google Scholar search reveals a team who have also explored using L-systems to model molecules; however, their paper is currently behind a
paywall. Based on the abstract, their system appears to be much more general than that presented here.[7]
Further applications of this idea could include modeling the structures of carbohydrates, generating exotic polypeptides, and exploring dendrimers.
Materials/Methods
The L-system used includes an alphabet V containing symbols for elements (C, H, O, S), bonds (_), and structural markers([,], +, -). The last two are
used to denote bond direction: + is a 90 degree turn to the left, - a turn to the right. The set V* consists of strings composed of symbols in V. The
axiom of an L-system,ω belongs to V*; here it is chosen to be a string representing a methane molecule:
ω = "C[_H][+_H][-_H][++_H]"
In other words, the axiom consists of a carbon atom with four branches, each containing a bond and a hydrogen atom. Different branches are offset in
space by different amounts of rotation.
The production rules P are defined mapping V* to V*. If s = x1x2.... is in V*, then P(s) = P(x1)P(x2)...
in V*. P is defined stochastically by specifying the possible outputs and the probabilities with which they occur:
| Code: | grammar = {
"H" : ["H","O_H","[C[+_H][-_H][_H]]"],
"C" : ["C"] ,
"O" : ["O", "S"]}
probability = {
"H": [0.8, 0.05, 0.15] ,
"O": [0.9, 0.1]}
|
Thus, the carbon backbone is left unchanged; the hydrogen atoms may be unchanged, replaced by a hydroxyl group, or converted into a sidebranch.
Oxygens are occaisonally converted into sulfur atoms, thus generating thiols.
Results
The program successfully generates non-cyclic alkanes, alcohols, ethers, and thiols. It does not appear to generate ungrammatical molecules. Current
visualization is not optimized, and as such, the turtle-based graphics are often not suited for larger molecules. However, molecules are also treated
as graphs internally, visualization of the graphs may be more useful, especially since atoms make a fixed number of bonds, and nodes can thus be
identified according to their degree.
A ground-up analysis of the production rules can also give a few insights. One observation is that maximum carbon backbone length increases as a
linear function of iteration number, n. At zero iterations, the only string in P0(ω) is ω which is a methane molecule with
backbone length 1. For each Pi(ω), consider the longest molecule; it has backbone length k. Select a hydrogen atom on each of the
terminal carbons and apply the methylating production rule; this is the longest backbone in Pi+1(ω) and it has length k+2
Inductively, the longest backbone in Pn(ω) is a chain of 2n+1 carbon atoms.
A similar argument can be applied to molecular weight. If we ignore thiols, the most massive molecules in any given iteration are the most heavily
branched; ie, those in which the methylating production rule has been applied to every available hydrogen atom. Applying this production rule to a
hydrogen adds 3 hydrogens and 1 carbon to the molecule; if the molecule has c carbon atoms and h hydrogen atoms, then applying the methylating rule to
all hydrogens adds h carbons and 3h hydrogens. The axiom, methane, starts with c=1 and h=4; thus in Pn(ω) the most densely branched
molecule is a C4n+1H4*3n
Discussion
The presented artificial chemistry is capable of producing molecules algorithmically. Conceptualizing molecules as strings of a formal language brings
them into a mathematically rich environment. [8] Furthermore, the ability to generate a collection of molecules could be used to explore the
statistical properties of the ensemble, eg, the distribution of molecular mass.
Other areas for further exploration include modeling of polysaccharides and the generation of exotic polypeptides. Different poly-glucose molecules
are distinguished by the degree of branching in their chains: “The main difference between glycogen and amylopectin is that glycogen is more highly
branched... branch points occur about every 10 residues in glycogen and about every 25 residues in amylopectin. In glycogen, the average chain length
is 13 glucose residues, and there are 12 layers of branching.” [5] This could be reproduced in silico by adjusting the probability parameters
describing branching frequency. Other branching molecules could also be generated. The sidechains of many amino acids offer bonding sites besides
their polypeptide backbone; examples of these could be algorithmically generated. Dendrimers also seem like a promising application. [6]
1) The Organic Builder: A Public Experiment in Artificial Chemistries and Self-Replication. Tim J. Hutton, February 21, 2007.</p>
2) Autocatalytic replication of polymers. Farmer, Kauffman, and Packard. Physica 220 (1986) 50-67</p>
3) A Graph-Based Toy Model of Chemistry. Benko, Flamm, and Stadler. August 19, 2002</p>
4) The Algorithmic Beauty of Plants.
Przemyslaw Prusinkiewicz and Aristid Lindenmayer, 1996. http://algorithmicbotany.org/
5) Biochemistry, p. 453 Campbell & Farrell Fifth ed.
6) http://en.wikipedia.org/wiki/Dendrimer
7) Model of chemistry molecule structure using L-system. Bin, Lin, & De Feng, Oct. 2010. DOI:10.1109/ICSTE.2010.5608782
8) Godel, Escher, Bach: An Eternal Golden Braid. Douglas Hofstadter
Attachment: examples.pdf (165kB)
This file has been downloaded 1266 times
|
|
|
woelen
Super Administrator
Posts: 7976
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
It seems to me that if you have built a generic engine for interpreting a grammar file, which gives the alphabet V of possible atoms, initial axiom
ω, extension rules and probablities, that you can easily add other types of molecules as well. This engine then can generate molecules as well.
It requires another level of programming though, not at specific rules but at a meta-level where you have rules about rules, rules about axioms and so
on.
It would be even more interesting if you could add rules from physical chemistry as well, such as spatial constraints, steric hindrance, exclusion of
unstable/non-existent groups like -O-O-O- .
|
|
|
mayko
International Hazard
Posts: 1218
Registered: 17-1-2013
Location: Carrboro, NC
Member Is Offline
Mood: anomalous (Euclid class)
|
|
Quote: Originally posted by woelen  | It seems to me that if you have built a generic engine for interpreting a grammar file, which gives the alphabet V of possible atoms, initial axiom
ω, extension rules and probablities, that you can easily add other types of molecules as well. This engine then can generate molecules as well.
It requires another level of programming though, not at specific rules but at a meta-level where you have rules about rules, rules about axioms and so
on.
It would be even more interesting if you could add rules from physical chemistry as well, such as spatial constraints, steric hindrance, exclusion of
unstable/non-existent groups like -O-O-O- . |
There is indeed a class called context-sensitive L-systems, which might be useful for comparing a given atom to its surroundings. It seems like a lot
of "neighbors of neighbors" checking though, which might be computationally intensive in larger molecules. The code I'm playing with is written for a
relatively specific kind of context-free system, and it would take a bit of programming to redo it, so I might not go that way.
One thing I was thinking about is using a Markov chain rather than just a probability table for the stochastic systems. That might add enough control
to avoid hyperperoxides. I also like the idea of writing empirically derived chemical reactions as production rules, tossing in some axioms and
growing a virtual pot full of reaction products 
|
|
|
mayko
International Hazard
Posts: 1218
Registered: 17-1-2013
Location: Carrboro, NC
Member Is Offline
Mood: anomalous (Euclid class)
|
|
I was walking around in the woods yesterday and I was looking at the cracks in the bark of the pine trees and got to thinking about how the patterns
looked like aromatic structures. One thought led to another and I started playing around with the idea of generating fractal aromatics with edge
replacement systems.
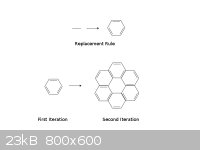
Here's a simple example. We take an edge (technically, the 1.5 bond in benzene) and replace each instance with a benzene ring. When this rule is
applied to a benzene molecule, each side of the benzene hexagon gets replaced with a benzene molecule, forming what I will call here hyperbenzene.
Notice that hyperbenzene contains two rings, an outer and an inner ring. I count 6 pi electrons in the inner ring and 18 in the outer ring; both of
these are 2 mod 4 so I would expect aromaticity from this molecule.
We can apply the rule again to get hyperhyperbenzene:
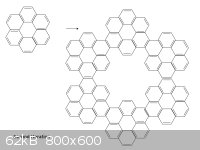
There is now an outer ring, an inner ring, and several smaller rings throughout. The smaller rings we have already seen to be aromatic; I count 66 pi
electrons in the outer ring and 30 in the inner ring, again both numbers are 2 mod 4.
I'm still working out a general proof that (hyper)^nbenzene is aromatic. I wonder what sort of NMR spectrum these molecules would have?
(derp, hit post too early...)
[Edited on 20-5-2013 by mayko]
|
|
|
Kapitan
Harmless
Posts: 9
Registered: 15-7-2013
Location: /dev/urandom
Member Is Offline
Mood: No Mood
|
|
Hmm. Well, on the larger one, the whole molecule has σ symmetry, so they'd be a singlet. The inner ones would also be a singlet for the same
reason. The off-center protons would be split by their neighbors, since they aren't symmetric, so they'd each yield a doublet. Every peak would have
the same integral. For the smaller molecule, I guess it'd just be one singlet, right?
That smaller one is called Coronene, and there's a proton and carbon spectrum for it on SDBS, if you're curious.
[Edited on 18-7-2013 by Kapitan]
|
|
|
mayko
International Hazard
Posts: 1218
Registered: 17-1-2013
Location: Carrboro, NC
Member Is Offline
Mood: anomalous (Euclid class)
|
|
I got a blurb about this paper in my NCBI daily digest:
Enumeration method for tree-like chemical compounds with benzene rings and naphthalene rings by breadth-first search order.
http://www.ncbi.nlm.nih.gov/pubmed/26932529
| Quote: |
BACKGROUND:
Drug discovery and design are important research fields in bioinformatics. Enumeration of chemical compounds is essential not only for the purpose,
but also for analysis of chemical space and structure elucidation. In our previous study, we developed enumeration methods BfsSimEnum and BfsMulEnum
for tree-like chemical compounds using a tree-structure to represent a chemical compound, which is limited to acyclic chemical compounds only.
RESULTS:
In this paper, we extend the methods, and develop BfsBenNaphEnum that can enumerate tree-like chemical compounds containing benzene rings and
naphthalene rings, which include benzene isomers and naphthalene isomers such as ortho, meta, and para, by treating a benzene ring as an atom with
valence six, instead of a ring of six carbon atoms, and treating a naphthalene ring as two benzene rings having a special bond. We compare our method
with MOLGEN 5.0, which is a well-known general purpose structure generator, to enumerate chemical structures from a set of chemical formulas in terms
of the number of enumerated structures and the computational time. The result suggests that our proposed method can reduce the computational time
efficiently.
CONCLUSIONS:
We propose the enumeration method BfsBenNaphEnum for tree-like chemical compounds containing benzene rings and naphthalene rings as cyclic structures.
BfsBenNaphEnum was from 50 times to 5,000,000 times faster than MOLGEN 5.0 for instances with 8 to 14 carbon atoms in our experiments.
|
al-khemie is not a terrorist organization
"Chemicals, chemicals... I need chemicals!" - George Hayduke
"Wubbalubba dub-dub!" - Rick Sanchez
|
|
|
mayko
International Hazard
Posts: 1218
Registered: 17-1-2013
Location: Carrboro, NC
Member Is Offline
Mood: anomalous (Euclid class)
|
|
These folks use a sort of edit-distance complexity measure to detect molecules that are "too complicated" to be specifically synthesized abiotically.
Still thinking it over but definitely interesting
Marshall, S. M., Mathis, C., Carrick, E., Keenan, G., Cooper, G. J. T., Graham, H., Craven, M., Gromski, P. S., Moore, D. G., Walker, S. I., &
Cronin, L. (2021). Identifying molecules as biosignatures with assembly theory and mass spectrometry. Nature Communications, 12(1), 1–9. https://doi.org/10.1038/s41467-021-23258-x
Attachment: Marshall et al. - 2021 - Identifying molecules as biosignatures with assembly theory and mass spectrometry.pdf (1.5MB)
This file has been downloaded 515 times
al-khemie is not a terrorist organization
"Chemicals, chemicals... I need chemicals!" - George Hayduke
"Wubbalubba dub-dub!" - Rick Sanchez
|
|
|
mayko
International Hazard
Posts: 1218
Registered: 17-1-2013
Location: Carrboro, NC
Member Is Offline
Mood: anomalous (Euclid class)
|
|
| Quote: Originally posted by mayko |
Notice that hyperbenzene contains two rings, an outer and an inner ring. I count 6 pi electrons in the inner ring and 18 in the outer ring; both of
these are 2 mod 4 so I would expect aromaticity from this molecule.
We can apply the rule again to get hyperhyperbenzene:
There is now an outer ring, an inner ring, and several smaller rings throughout. The smaller rings we have already seen to be aromatic; I count 66 pi
electrons in the outer ring and 30 in the inner ring, again both numbers are 2 mod 4.
I'm still working out a general proof that (hyper)^nbenzene is aromatic. I wonder what sort of NMR spectrum these molecules would have?
|
I ran across this article which looks at the idea of molecule-wide aromatic effects ("hyperaromaticity") in fused-ring systems like this. Apparently
the consensus from computational/NMR studies is that the aromaticity is mostly confined in local, six-sided rings and any contribution of
molecule-wide delocalization is small. Oh well 
Buttrick, J. C., & King, B. T. (2017). Kekulenes, cycloarenes, and heterocycloarenes: Addressing electronic structure and aromaticity through
experiments and calculations. Chemical Society Reviews, 46(1), 7–20. https://doi.org/10.1039/c6cs00174b
Attachment: Buttrick, King - 2017 - Kekulenes, cycloarenes, and heterocycloarenes.pdf (2.5MB)
This file has been downloaded 247 times
OTOH, this computational study predicted opposing ring currents on the inner and outer perimeters when a similar molecule is placed in a magnetic
field, which is kinda cool:
Monaco, G., Viglione, R. G., Zanasi, R., & Fowler, P. W. (2006). Designing ring-current patterns: [10,5]-coronene, a circulene with inverted rim
and hub currents. The Journal of Physical Chemistry. A, 110(23), 7447–7452. https://doi.org/10.1021/jp0600559
Attachment: Monaco et al- 2006 - Designing ring-current patterns .pdf (335kB)
This file has been downloaded 212 times
I ran into this stuff via the paper that came out last year on the synthesis of inifinitene, in which twelve rings are joined in a figure-eight loop.
Apparently the untwisted isomer hasn't been synthesized yet!
Krzeszewski, M., Ito, H., & Itami, K. (2022). Infinitene: A Helically Twisted Figure-Eight [12]Circulene Topoisomer. Journal of the American
Chemical Society, 144(2), 862?871. https://doi.org/10.1021/jacs.1c10807
Attachment: Krzeszewski - Infinitene A Helically Twisted Figure-Eight 12 Circulene Topoisomer.pdf (5.1MB)
This file has been downloaded 277 times
al-khemie is not a terrorist organization
"Chemicals, chemicals... I need chemicals!" - George Hayduke
"Wubbalubba dub-dub!" - Rick Sanchez
|
|
|
|










