| Pages:
1
2
3
..
5 |
madscientist
National Hazard

Posts: 962
Registered: 19-5-2002
Location: American Midwest
Member Is Offline
Mood: pyrophoric
|
|
Catechol preparation
I have been interested in preparing Benzene-1,2-diol (catechol) for some time. Here's the route of synthesis that I think would be viable for the
amateur chemist.
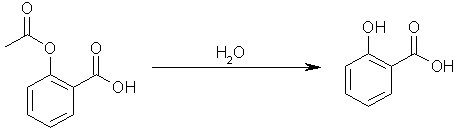
Step one: hydrolyze acetylsalicylic acid to yield salicylic acid. Acetic acid will be another product of the reaction.
Step two: react salicylic acid with carbamide to yield salicylamide. Ammonia and carbon dioxide gas will be evolved by this reaction.
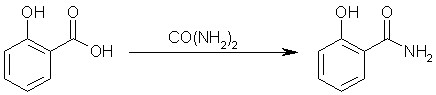
Step three: perform a Hofmann elimination on salicylamide with calcium hypochlorite to yield 1-hydroxyl-2-aminobenzene. Carbon dioxide will be
evolved, and calcium chloride will form as well.
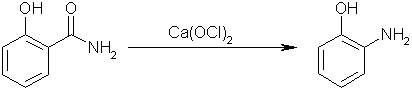
Step four: add sodium nitrite to 1-hydroxyl-2-aminobenzene to yield the monosodium salt of Benzene-1,2-diol. This works by the acidic hydroxyl group
reacting with the sodium nitrite to form nitrous acid, which then performs a diazotation on the amino group, yielding the desired hydroxyl group.
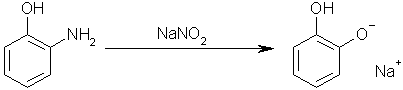
Step five: add hydrochloric acid to yield Benzene-1,2-diol. Sodium chloride will obviously be the other product.
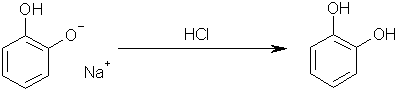
Step six: boil off all water, add toluene in which the Benzene-1,2-diol will dissolve; pour the toluene through a filter (to catch any sodium chloride
crystals that may have broken off into the toluene); distill off the toluene, leaving Benzene-1,2-diol crystals.
I weep at the sight of flaming acetic anhydride.
|
|
|
The_Cutting_Crew
Harmless
Posts: 1
Registered: 13-11-2002
Location: *
Member Is Offline
Mood: No Mood
|
|
pyrocatechol
It`s a good base-stuff for stabilising-agents than
3,5-di-tertbutyl-1,2-benzenediol or to build other
X-nitro-catchol-derivates.
Have You a idea whats the synthesis-way when
adding HCl to
hydro-sodium-pyrocatechol-3,5-disulfonate ?
C6H4O8S2Na2 + HCl ???

|
|
|
IodineForLunch
Harmless
Posts: 34
Registered: 28-8-2002
Location: NJ
Member Is Offline
Mood: bored
|
|
Are you on drugs?
|
|
|
madscientist
National Hazard
Posts: 962
Registered: 19-5-2002
Location: American Midwest
Member Is Offline
Mood: pyrophoric
|
|
Adding HCl to the molecule you shared a diagram of should yield the free acid of that salt: 4,5-Dihydroxy-benzene-1,3-disulfonic acid.
Perhaps I misunderstood what you said? 
I weep at the sight of flaming acetic anhydride.
|
|
|
KABOOOM(pyrojustforfun)
Hazard to Others
Posts: 254
Registered: 12-10-2002
Location: Iran (pseudoislamic dictatorship of)
Member Is Offline
Mood: exuviating!
|
|
I think this method would be easier:
dissolve phenol in excess conc H2SO4 at low temperatures to get ortho-phenolsulfonic acid (high temp leads into
parapheolsufonic acid)
add exact calculated amount of NaOH needed, then gently warm it to evaporate all the water so that a solid stuff remains.
attack a condenser to the flask, and heat to 240°C(bp of catechol) and keep on this temperature until no more catechol is distilled.
C6H5OH + H2SO4 => HOC6H4SO3H + H2O
HOC6H4SO3H + NaOH => NaHSO3 + C6H4(OH)2
|
|
|
madscientist
National Hazard
Posts: 962
Registered: 19-5-2002
Location: American Midwest
Member Is Offline
Mood: pyrophoric
|
|
Are you sure that reacting sodium hydroxide with an aromatic sulfonic acid causes the oxidation state of the sulfur to drop? Seems mighty strange to
me.
I thought of another, more economical idea for synthesizing catechol (since acetylsalicylic acid isn't exactly inexpensive). Toluolsulfonic acid can
be hydrated to yield toluene and sulfuric acid by heating with water. The ortho form of toluolsulfonic acid is prepared by reacting concentrated
sulfuric acid with toluene. So, perhaps 2-hydroxyltoluene could be prepared by reacting concentrated sulfuric acid with toluene; purifying the
toluolsulfonic acid crystals; heating with hydrogen peroxide; and then extracting and purifying the 2-hydroxyltoluene. The 2-hydroxyltoluene would be
oxidized, then reacted with urea, then treated with calcium hypochlorite, and then diazotized with nitrous acid to yield catechol.
I weep at the sight of flaming acetic anhydride.
|
|
|
Marvin
National Hazard
Posts: 995
Registered: 13-10-2002
Member Is Offline
Mood: No Mood
|
|
Concentrated sulphuric acid is actually half decent oxidising agent, the method stated is not disimilar to one of the methods for making phenol. Conc
sulphuric with a catalyst will even oxidise napthalene to pthallic acid.
I'm very bothered by the idea of using hoffman rearangement to make an aryl amine, do we have any reliable information of this being done?
|
|
|
KABOOOM(pyrojustforfun)
Hazard to Others
Posts: 254
Registered: 12-10-2002
Location: Iran (pseudoislamic dictatorship of)
Member Is Offline
Mood: exuviating!
|
|
madscientist the method I mentioned was right except the mistake about temperature
extracted from my "THE CONDENCED CHEMICAL DICTIONARY" Revised by GESSNER-G.HAWLEY :
pyrocatechol (ortho-dihydroxybenzene; catechol)
C6H4(OH)2.
Properties: Colorless crystals; discolors to brown on
exposure to air and light, espesially when moist; sp.
gr. 1.371; m.p. 104°C; b.p. 245°C, sublimes; soluble
in water, alcohol, ether, benzene and chloroform,
also in pyridine and aqueous alkaline solutions.
Combustible. Flash point 261°F (127°C) (C.C).
Derivation: (a) By fusion of ortho-phenolsulfonic
acid with caustic potash at 350°C. (b) By heating
guaiacol with hydroiodic acid.
Grades: Technical; C.P.; resublimed.
Containers: 25- to 200-lb drums.
Hazard: Strong irritiant. Toxic. Tolerance, 5 ppm in
air.
Uses: Antiseptic; photography; dyestuffs; electro-
plating; specialty inks; antioxidants and light stabil-
izers; organic synthesis.
I also have "Dictionary of Chemistry" (by David William Arthur Sharp) that mentions melting ortho-benzene-disulphonic acid with NaOH
|
|
|
Ritter
Hazard to Others
Posts: 370
Registered: 20-6-2008
Location: Earth
Member Is Offline
Mood: Curious
|
|
Catechol (along with hydroquinone) are made industrially via hydrogen peroxide oxidation of phenol.
Ritter
=============================
\"The production of too many useful things results in too many useless people.\"
Karl Marx
|
|
|
stoichiometric_steve
National Hazard
Posts: 819
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
why not just buy the stuff, it's dead cheap - 250g 30 USD...
|
|
|
YT2095
International Hazard
Posts: 1091
Registered: 31-5-2003
Location: Just left of Europe and down a bit.
Member Is Offline
Mood: within Nominal Parameters
|
|
madscientist:
what are using as the medium to perform these reactions in?
it looks like a very Simple straightforwards procedure and one I would like to try myself even though I already have Catechol.
I would be starting from the Salicylic acid stage, and this doesn`t dissolve in water very well.
\"In a world full of wonders mankind has managed to invent boredom\" - Death
Twinkies don\'t have a shelf life. They have a half-life! -Caine (a friend of mine)
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
That hydroxide fusion cannot be performed in usual glassware, it will etch any pyrex within minutes...
A dakin reaction on salicylaldheyde should yield the deired compound in ratehr high yields. Salicylaldehyde can be produced from phenol in 84% yields
via Mg(OCH3)/(H2CO)n formylation, or by a riemer-tieman formylation.
You would have to compare with the direct H2O2 oxidation of phenol, although the two step preparation above yields only one isomer.
For the amide formation, you might better be off forming the ester and reacting it with alcoholic ammonia, a bit longer but less decomposition
products. I know the hofmann degradation works for nitrobenzamide for sure, but don't know how well it would perform here. Check the litterature.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
A cool and stupid easy way of making chloramides for the hofmann degradation involves stirring the amide with TCCA in MeOH and allowing the cyanuric
acid to percipitate out as the amide is chlorinated. I tried this with urea as a substrate and it worked just as described in the article attached
below.
I think though, your method would not be very efficient as I would worry about ring chlorination. Phenols have a pretty activated nucleus, and the
conditions for the classic Hofmann degradation would certainly halogenate it. Although maybe your method with straight calcium hypochlorite instead
of the classic Br2 and then NaOH/Heat may not suffer this problem.
Attachment: N-Chloroamides_via_TCCA.pdf (113kB)
This file has been downloaded 2941 times
|
|
|
Ritter
Hazard to Others
Posts: 370
Registered: 20-6-2008
Location: Earth
Member Is Offline
Mood: Curious
|
|
| Quote: | Originally posted by stoichiometric_steve
why not just buy the stuff, it's dead cheap - 250g 30 USD... |
This is the same question I ask about subscribers who post elaborate & expensive schemes to make very inexpensive chemicals. In most cases it is
intellectual curiosdity rather than economics, I would guess.
Ritter
=============================
\"The production of too many useful things results in too many useless people.\"
Karl Marx
|
|
|
ScienceSquirrel
International Hazard
Posts: 1863
Registered: 18-6-2008
Location: Brittany
Member Is Offline
Mood: Dogs are pets but cats are little furry humans with four feet and self determination! 
|
|
I think you will find that the easiest way from OTC chemicals is;
1) Aspirin to salicylic acid
2) Salicyclic acid to phenol-2-sulphonic acid with concentrated sulphuric acid ( ipso substitution )
3) Fusion with caustic potash to yield catechol
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
IMHO, hydroxide fusion isn't avery practical reaction: it can't be done in conventional borosilicate glassware without causing excessive damage (risk
or breaking the beaker/flask once the glassware has been significantly etched), and melting an alkali hydroxide in a crucible or such is very
hazardous.
It would be great if there was a way of performing this reaction in less extreme ways.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
ScienceSquirrel
International Hazard
Posts: 1863
Registered: 18-6-2008
Location: Brittany
Member Is Offline
Mood: Dogs are pets but cats are little furry humans with four feet and self determination!
|
|
Here is an elegant method using the Dakin reaction
Phenol -> Salicylaldehyde - > Catechol
Maybe someone could post the full paper
One-pot synthesis of substituted catechols from the corresponding phenols
Tetrahedron Letters, Volume 46, Issue 19, 9 May 2005, Pages 3357-3358
|
|
|
FreedomFighter
Harmless
Posts: 8
Registered: 21-9-2007
Location: Hispania
Member Is Offline
Mood: No Mood
|
|
Take a look at US patent 4465864, may be you can apply it to phenol and get pyrocatechol.
|
|
|
Mush
National Hazard
Posts: 632
Registered: 27-12-2008
Member Is Offline
Mood: No Mood
|
|
Methyl salicylate -> salicylamide
US2570502
Problem in the diazotation:
"Due to the adjacency of the amino and hydroxyl groups, 2-aminophenol readily forms heterocycles."
Reference
Another route:
Benzene (nitration)-> nitrobenze (reduction) ->
aniline (diazotation) ->phenol (Reimer-Tiemann formylation)
mostly o-salicylaldehyde (Dakin Reaction)-> pyrocatechol
Nitrobenzene
Vogel, Practical Organic Chemistry, 5 th ed., p 854
This preparation should be conducted in an efficient fume cupboard.
Place 50 g (35 mL, c. 0.5 mol)of concentrated nitric acid in a 500-mL round-bottomed flask,
and add, in portions with shaking, 74 g (40 mL) of concentrated sulfuric acid. Keep the
mixture cool during the addition by immersing the flak in cold water. Place a thermometer
(110 °C range) in the acid mixture. Introduce 26 g (30 mL, 0.33 mol of benzene (CAUTION)
in portions of 2 - 3 mL; shake the flask well to ensure thorough mixing, after each addition
of benzene. Do not allow the temperature to rise over 55 °C; immerse the flask, if necessary,
in cold water or in ice water. When all the benzene has been added, fit a reflux condenser
to the flask and heat it in a water bath maintained at 60 °C (but not appreciably higher)
for 40 - 45 minutes; remove the flask from time to time and shake it vigorously to ensure
good mixing of the immiscible layers. Pour the contents of the flask into about 500 mL of
cold water in a beaker, stir the mixture well in order to wash out as much acid as possible
from the nitrobenzene and allow to stand. When the nitrobenzene has settled to the bottom,
pour of the acid liquor as completely as possible, and transfer the residual liquid to a
separatory funnel. Run off the lower layer of nitrobenzene and reject the upper aqueous
layer; return the nitrobenzene to the separatory funnel and shake it vigorously with about
50 mL of water. Separate the nitrobenzene as completely as possible and rin it into a small
conical flask containing about 5 g of anhydrous calcium chloride. If the nitrobenzene does
not become clear because of the presence of emulsified water, warm the mixture, with shaking,
for a short period on a water bath; the cloudiness will soon disappear. Filter the cold product
through a small fluted filter paper into a small (50- or 100-mL)distilling flask and attach
a still-head and air condenser. Heat the flask on a ceramic-centred wire gauze or preferably
in an air bath, and collect the fraction which boils at 206 - 211 °C. (1). Do not distill
quite to dryness nor allow the temperature to rise above 214 °C, for there may be a residue
of m-dinitrobenzene or higher nitro compounds and an explosion may result. The yield of
nitrobenzene is 35 g (85 %). Pure nitrobenzene is a clear, pale yellow liquid, B.P. = 210 °C.
(1) Nitrobenzene is appreciably toxic and its vapour should not be allowed to escape into
the atmosphere of the laboratory. Site the distillation apparatus in a fume cupboard, use
the receiver assembly illustrated in fig. 2.98, and attach to the outlet of the receiver
adapter a piece of rubber tubing leading to the extraction system. The liquid is also a
skin poison, if accidentally spilled on the skin, it should be removed by washing with a
little methylated spirit, followed by soap and warm water.
Nitrobenzene Synthesis (alternative)
Prepare a mixture of 82 mL of 95-100% sulfuric acid and 71 mL of 70% nitric acid in a 500-mL
flask. Stir well and allow the mixture to cool to room temperature in a cold water bath.
Gradually add 57 mL of benzene to the acid with frequent shaking. If the temperature rises
above 50-60 °C during the benzene addition, stop adding benzene and cool the flask in a
cold water bath until the temperature has lowered. After all of the benzene has been added,
reflux the flask in a water bath at 60 °C for 1 hour. The temperature of the water bath
should be 60 °C, not the contents of the flask. Shake the flask frequently during reflux.
After heating, allow the flask to cool, two layers should form. Transfer the contents
to a separatory funnel and drain off the bottom layer of sulfuric and nitric acids; the
top layer contains the nitrobenzene. The bottom layer can be disposed of. The nitrobenzene
is then vigorously shaken in the separatory funnel several times with water. After each shaking,
allow the layers to separate, the nitrobenzene will now be the bottom layer, dispose of the
top water layer. After washing, place the nitrobenzene in a dry Erlenmeyer flask with some
calcium chloride. Heat this flask on a steam bath, it will first be milky, then it will
go clear, stop when it is clear. The nitrobenzene is now purified by simple distillation.
Yield is about 60-70 g.
Aniline
Vogel, Practical Organic Chemistry, 5 th ed., p 892
Into a 500-mL round bottomed flask equipped with a reflux condenser place 25 g (21 mL, 0.25 mol) of nitrobenzene and 45 g (0.38 mol) of granulated
tin. Measure out 100 mL of concentrated hydrochloric acid. Pour about 15 mL of this acid down the condenser and shake the contents of the flask
steadily. The mixture becomes warm and before long the reaction should be quite vigorous; if it boils very vigorously, moderate the reduction somewhat
by temporarily immersing the flask in cold water. When the initial reaction slackens of its own accord, pour another 15 mL of hydrochloric acid down
the condenser, shake the flask steadily to ensure thorough mixing and cool again if the reduction becomes too violent. Do not cool more than is
necessary to keep the reaction under control; keep the mixture well shaken. Proceed in this way until all 100 mL of acid has been added. Finally heat
the mixture on a boiling water bath for 30 - 60 minutes, i.e. until the odour of nitrobenzene is no longer perceptible and a few drops of the reaction
mixture when diluted with water yield a perfectly clear solution. During the course of the reduction, particularly during the cooling, aniline
chlorostannate may separate as a white or yellow crystalline complex. Cool the reaction mixture to room temperature and add gradually a solution of 75
g of sodium hydroxide in 125 mL of water; if the reaction mixture boils during the addition of alkali, cool again. The hydroxide of tin which is first
precipitated should all dissolve and the solution should be strongly alkaline: the aniline separates as an oil. Equip the flask for steam
distillation, and pass steam into the warm mixture until, after the distillate has ceased to pass over as a turbid liquid, a further 120 mL of clear
liquid are collected. Since aniline is appreciably soluble (c. 3 %) in water, it must be 'salted out' by saturating the distillate with salt. Use
about 20 g of commercial salt for each 100 mL of liquid. Transfer the distillate, saturated with salt, to a separatory funnel, add about 40 mL of
ether and shake to ensure intimate mixing of the solution and the ether; relive the pressure within the funnel by momentarily lifting the stopper.
(All flames in the vicinity must be extinguished during the extraction ... :-) you must be joking ...). Allow the two layers to separate; run off the
lower aqueous layer into a beaker, and pour the ethereal layer through the mounth of the funnel into a 200-mL flask. Return the aqueous solution to
the funnel and extract with a further 40 mL of ether. Proceed as before, and pour the ethereal extract into the flask. Dry the combined ethereal
solutions with a few grams of anhydrous potassium carbonate (1): shake the well-stoppered flask for several minutes. Filter the ethereal solution
through a fluted filter paper and remove the ether by flash distillation, using a 50-ml round-bottomed flask to which has been added a few boiling
chips. Since ether is extremely volatile and also highly flammable, the flask must be heated by means of an electrically heated water bath. When all
the ethereal solution has been introduced into the flask, and no more ether distilson the boiling water bath, run out the water from the condenser,
and distil the aniline either by direct heating over a wire gauze or, preferably, using an air bath. A small quantity of ether may pass over during
the early part of the distillation; it is therefore advisable to interpose a uralite board between the receiver and the flame. Collect the fraction
B.P. 180 - 184 °C, in a weighed conical flask. The yield of aniline is 18 g (97 %).
Phenol (with diazotation)
Attachment: Organikum (2001), p637-638 Phenol preparation.rar (250kB)
This file has been downloaded 1092 times
Simply heat up your reaction mixture till
nitrogen formation subside to get phenol.
Salicylaldehyde
Reimer-Tiemann formylation process for producing aldehydes
(US4755613)
EXAMPLE 1
100.cm3 of chloroform, 9.4 g of phenol, 20 g of anhydrous sodium hydroxide and 3.6 cm3
of water are placed in a 250 cm3 reactor provided with a coolant, a mechanical stirrer
and a thermometer, and the reaction medium temperature is thermostatically maintained
at 50° C. These conditions of the initial mixture correspond to a hydration rate of
0.4 moles of water per mole of sodium hydroxide (2 moles of water per mole of initial
compound) and to 12.6 times as much chloroform as initial compound (expressed in moles).
Thereupon the temperature of the reaction medium is raised to 58° C. for one hour.
Next 12 g of sodium hydroxide in powdered form are progressively added over a period of
two hours, the temperature being kept constant at 58° C. The reaction proceeds for 1
hour. At the end of the reaction, the initial phenol has completely disappeared.
The residual chloroform is recovered and recycled. The mixture of aldehydes is
obtained in sodium form. It is neutralized until a neutral brine is obtained.
The salicylaldehyde can be recovered in conventional manner at a yield close to 77% (9.4 g)
with respect to the initial phenol by carrier vapor distillation or by ether extraction.
The p-hydroxybenzaldehyde is recovered at a yield of 17% (2 g) by ether extraction from
the acidified residual brine to pH of 1.
Catechol (pyrocatechol)
Organic Syntheses, Coll. Vol. 1, p.149 (1941); Vol. 3, p.28 (1923).
To a solution of 122 g. (1 mole) of pure salicylaldehyde (Note 1) in 1000 cc. of normal
sodium hydroxide solution at room temperature, is added 1420 g. (1.2 moles) of 3 per cent
hydrogen peroxide. The mixture darkens slightly in color and the temperature rises to 45–50°.
The solution is allowed to stand for fifteen to twenty hours, whereupon a few drops of
acetic acid are added in order to neutralize any excess alkali, and the solution evaporated
to complete dryness on the water bath under reduced pressure.
The solid residue is finely crushed and warmed nearly to boiling with 500 cc. of toluene;
the mixture is then poured into the folded filter paper of an extraction apparatus (p. 375)
and extracted with boiling toluene for five hours. The toluene is allowed to cool and is
decanted from the catechol, which crystallizes out. The insoluble material is again ground
up and extracted in the apparatus with the decanted toluene. The combined product,
weighing 70–76 g., consists of light brown plates melting at 104°, and is thus pure enough
for many purposes. A further 6–12 g. of catechol can be obtained on distilling off the bulk
of the toluene from the mother liquor. In order to obtain an entirely pure product, the
crude catechol should be distilled under reduced pressure, when it passes over entirely at
119–121° /10 mm. (or 113–115° /8 mm.), and the distillate recrystallized from about five
times its weight of toluene. In this way, colorless plates melting at 104–105° are obtained.
The yield of the purified product is 76–80 g. (69–73 per cent of the theoretical amount) (Note 2).
Notes
1. A considerably lower yield is obtained (50 per cent or less) if technical salicylaldehyde
(not purified through the bisulfite compound) is employed.
2. The procedure described is applicable to almost all hydroxyaldehydes in which the hydroxyl
and carbonyl groups occupy ortho or para positions relatively to each other;1 in the latter
case derivatives of hydroquinone are produced. When the hydroxyl and carbonyl groups occupy
the meta position with respect to each other, no reaction takes place, as is also the case
with certain ortho and para compounds containing nitro groups and iodine atoms.
o-Hydroxyacetophenone and p-hydroxyacetophenone are also capable of yielding catechol
and hydroquinone, respectively, under the above conditions.
3. Catechol may also be produced from salicylaldehyde by the use of certain derivatives
of hydrogen peroxide, such as persulfates or sodium peroxide, but the method is far less convenient.
[Edited on 19-8-2009 by Mush]
|
|
|
JohnWW
International Hazard
Posts: 2849
Registered: 27-7-2004
Location: New Zealand
Member Is Offline
Mood: No Mood
|
|
- try instead http://www.pat2pdf.org/patents/pat2570502.pdf
- See the References section for the complete book by Vogel.
- try instead http://www.pat2pdf.org/patents/pat4755613.pdf
|
|
|
unome
Hazard to Others
Posts: 134
Registered: 17-10-2009
Member Is Offline
Mood: No Mood
|
|
Why would diazotization be necessary? There are several papers where p/m/o-amino groups have been removed during heating of compounds with syrupy
phosphoric acid (granted most are involving modified Fischer cyclization), but suffice to say, the relevant amino groups have been removed in
essentially quantitive yield.
If that were the case, the only remaining issue I can see is the possibility of dimer/trimer formation, plus the fact I haven't seen any mention of a
Hoffman rearrangement of 2-hydroxybenzamide in any literature (which might lead suspicious minds to suspect there are workup problems of one form or
another)...
It would be nice if the Reimer-Tiemann could be avoided, but if not, then I really want catechol simply because of the ease of forming the
benzodioxole without needing inert atmospheres/etc.
PS I am in Australia and there is no really cheap source of photochems, the one decent source is overly inquisitive, prone to reporting suspicious
purchases and not at all pleasant to deal with. As for importing ANY chemical, no chance in hell.
|
|
|
unome
Hazard to Others
Posts: 134
Registered: 17-10-2009
Member Is Offline
Mood: No Mood
|
|
Have to double-post (must have missed the deadline) but for anyone here who (1) speaks German; and (2) has access to Ber. this reference apparently
(according to page 291 of Organic Reactions, Vol 3 - this sites library) details the formation of 2-hydroxyaniline from salicylamide via an
intermediate 4,5-benzoxazole (??)
Graebe and Rostowzev, Ber. 35, 2747 (1902)
Details (and especially a translation) would be gratefully accepted
PS The relevant article is now attached (with many thanks to Java/Solo) if any of the German speakers could help out, the problem might well me
capable of being solved
[Edited on 24-11-2009 by unome]
Attachment: phpuFO2fA (401kB)
This file has been downloaded 1480 times
|
|
|
something_sinister
Harmless
Posts: 1
Registered: 21-2-2010
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by unome  |
PS I am in Australia and there is no really cheap source of photochems, the one decent source is overly inquisitive, prone to reporting suspicious
purchases and not at all pleasant to deal with. As for importing ANY chemical, no chance in hell. |
http://www.vanbar.com.au, is an excellent Australian photochemical site. I just got my order of mercuric chloride, sodium bromide and potassium
iodide today. Catechol per 100gm $60. suspicious/no chance in hell... Your just not looking hard enough mate. Australia may seem like one of the most
restricted places on earth, but theres still hope for those chemists who persevere.
|
|
|
Panache
International Hazard
Posts: 1290
Registered: 18-10-2007
Member Is Offline
Mood: Instead of being my deliverance, she had a resemblance to a Kat named Frankenstein
|
|
Eugenol will demethylate to catechol in a NaCl/AlCl3 melt (i think the proportions for the eutetic are 3:1) at ~175C. Yields are terrible, around 20%
however the reagants are cheap (assuming you make your own AlCl3, purity of the Alcl3 is not paramount). Also the reaction is quick and simple...form
the eutetic in a beaker raise to 200C, drip in the eugenol as quickly as possible without bombing out the temperature (and whilst standing back as it
hisses and fizzes), once the eugenol has been added, leave at temperature for a few minutes then pour the vile tarry mess into an excess of ice water.
The fun really begins now as the workup is tedious.
It will appear to run unsuccessfully many times and suddenly the tarry vile mess will almosty seem pure, in stark contrast to most runs, the reason
for this is luck (or maybe Jesus helping out). Interestingly although far more pleasant to workup the yeilds are not any better on these hail mary
runs, and once the workup is grasped the reaction runs well.
Vac distill the eugenol prior to use as this tends to render the workup simplier, i am assuming the myriad of natural poroducts present in small
quantites in eugenol wreac havoc with the cataechol formed generating messy complexes with the aluminium.
i just made up a chemistry joke
q.did you hear about the chemist demethylating eugenol out in the snow
a;he caught a cold! (catecol, LMOA, ROFL, LOL, Ha!, teet)
|
|
|
peach
Bon Vivant
Posts: 1428
Registered: 14-11-2008
Member Is Offline
Mood: No Mood
|
|
Interesting about a melt and the eutectic point. Personally, I've never had any luck with the stuff even when simply using AlCl3 in a polar aprotic or
none polar solvent. Usually ends up with a nice tarry mess. And what's worse, it starts forming the second the first drop hits the solution (I can
see the solution darkening to 'that color' we all know and love).
I've even tried AlI3 in none polar solvent with a PTC under argon. It yielded a quantity of thick, lightly green colored resinous material that became
considerably tough after cooling in the fridge (I could leave the flask on it's side for a few hours and it wouldn't move). When back at room
temperature, it'd flow a few mm if left on it's side for hours. The amount of product, this time, was more in line with the quantity of substrate
used. Importantly, it wasn't the color of a smokers lungs (my poor, poor lungs). If I poked at the resin like material, I could leave impressions in
it's surface and it wouldn't stick to the poking instrument. Over time, exposed to the atmosphere, it appeared to darken to a more greeny brown color.
I also tried said AlI3 method using DCM, producing an odd result that was damn near IMPOSSIBLE to filter. By that I mean, I had to leave it for two
days to collect tens of mls. I could swirl it and scrape at the filter paper all I liked. Nothing. And the filtrate seemed to refuse to boil at room
temperature when down around 40mBar, despite being full of DCM. I abandoned the filtrate but left it to stand in the sunlight, exposed to the
atmosphere. It gradually turned almost black over a few days. Hmmm... interesting!
When I was trying to demethylate with AlCl3, I noticed what I am sure where HCl fumes slowly leaving the flask on addition. I am sure they were HCl
due to the acrid nature, smell, taste and brass around them turning green. This may have been water in the substrate. I'm fairly sure this substrate
has been vacuum distilled prior to me getting hold of it, and it hasn't been left open. So it's unlikely to have much water in it. And the amount of
fuming seemed out of proportion to any realistic water content of the oil. I also agree that using raw oil is probably a bad idea, since caryophyllene
(one of it's other components) features a few double bonds, which will likely be getting intimately involved with the acid, in a bad way (eugenol is
enough of a bastard child on it's own). I tried A/B extracting the raw oil and received a product that was still a significantly brown/orange color
with a complex aroma. The distillate is near colorless and has a very simple aroma.
On hydrolysing AlCl3 runs, there would inevitably be a large amount of HCl produced, far more than when adding the eugenol. This was done in an ice
bath with ice cooled water. The aluminum would also go into solution in the aqueous phase.
AlI3 runs were different. Here, adding the eugenol produced significantly more fuming. I also suspect this was HI. It would immediately begin turning
the solution opaque. On heating, the solution would immediately thicken and begin turning gray. After ten or so minutes at only the reflux point of
DCM, it would form a solid grey sludge and cake up, jamming a 70mm odd sized stir bar in an erlenmeyer. The cakeyness (advanced SI terminology  ) would actually form a skin and form 'crevices' through it's self, like an over
baked milky pudding. At hydrolysis, absolutely nothing would happen. No fuming, no breakdown of the cake. I had to warm the solution to just above
room temperature. Again, no evolution of gas. But the cake would then breakdown into fine grains of aluminum. ) would actually form a skin and form 'crevices' through it's self, like an over
baked milky pudding. At hydrolysis, absolutely nothing would happen. No fuming, no breakdown of the cake. I had to warm the solution to just above
room temperature. Again, no evolution of gas. But the cake would then breakdown into fine grains of aluminum.
I've tried AlCl3 in DCM in a salt/ice bath, same tarryness on addition.
As I say, the AlI3 methods are stupidly difficult to filter. I can pick the filter paper up and hold it there for a minute and nothing will come
through. Same for swirling and stirring. It seems, ironically, easier when using a higher BP solvent, and virtually impossible when using something as
free flowing as DCM.
With regards to AlCl3 purity, I've bought CP grade before and it's literally been dirt brown. After leaving it to sit in some DCM for a few weeks, I
just about managed to clean most of it off. Some of the crystals were also tinted yellow. That's suggesting the aluminum was contaminated with iron.
How on earth it got that dirty is something I'm still not sure of. It looked like they'd scraped the aluminum up off the sides of the electrolytic
cells.
Intriguing my dear Watsons, intriguiging.
As for synthesizing the stuff through a method as long as the original post... darn! Time to switch your developing solutions if it's for anything
other than the fun of playing. 
If you're considering something featuring as many stage as that, you could also look to guaiacol, which will probably be easy to demethylate to
catechol and occurs in both the plant guaiacum and wood creasote (tar). Interesting facts about each... the wood from guaiacum is the hardest known.
It lives around the Carribean and northern South America. Don't know if it's native to the land of girls, sunshine, beaches and positivity but the
climate is about right. Not worth growing it since it's a slow grower. Wood creosote is used to preserve wood furniture or other timber in the garden.
I'm pretty sure you could find a gigantic 25l container of it if you ask at the timber yards. They may try selling you expensive, proprietary, brand
name timber solutions. Answers;
"I like the smell of creosote in the morning"
"My [boss, strange brother, farther, alter ego, someone who knows lots about wood] sent me here and, I have no idea why, but he specifically demanded
wood creosote"
"I'm making my own photodeveloper" (wood creosote actually changes color when exposed to light I think)
You want WOOD creosote, not coal creosote. Coal creosote doesn't have the guaiacol in it and it's full of things like PAHs (a primary carcinogen found
in cigarette smoke). It does make nice smelling soap however.
The number of steps involved in what you're thinking of is already a good few. If you were going on from there to something else via a few more
stages, that would be one heavy and involved workload, possibly resulting in horrendously poor yields.
[edit]I'm looking at a 25l container of creosote now and it's about $95 AUD. Also, be advised that creosote is usually used by people with experience
when it comes to timber, so making out like you're the one using it in the garden and then having no idea what you're talking about when they ask you
about timber may encourage them to question your ability to use it in the first place. e.g. "What's your stock? (Good sir, what wood is it that you
will be using this fine product upon?" "Erm.........."[/edit]
[Edited on 20-6-2010 by peach]
|
|
|
| Pages:
1
2
3
..
5 |
|










