| Pages:
1
2
3
4
5 |
bbartlog
International Hazard

Posts: 1139
Registered: 27-8-2009
Location: Unmoored in time
Member Is Offline
Mood: No Mood
|
|
Ah, you're right. My bad. Fair enough; I guess you can get 3-acetylsalicylic acid that way. I remain skeptical about this being a usable precursor for
catechol, though.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
The only mention related to the Fries rearrangement of aspirin that I could find is in some old article in a Romanian journal. HClO4 is used for in
situ O-acylation and subsequent Fries rearrangement of various phenols, salicylic acid included. Yields are low and I doubt they would be much better
by starting from aspirin directly. Unfortunately, the journal only has electronic issues starting from 2006, so unless you get in in a library, the CA
abstract is the only info available:
| Quote: | Fries rearrangement. Leonte, Mircea; Beschia, Magda; Pascaru, Elvira; Stoica, Maria. Polytech. Inst., Galati, Rom. Studia Universitatis
Babes-Bolyai, Chemia (1963), 8(1), 291-6. CODEN: SUBCAB ISSN: 1224-7154. Journal language unavailable. CAN 61:68907 AN 1964:468907
CAPLUS
Abstract:
Starting from the hypothesis that the strength of the acid catalysts plays an essential role in the initiation of the Fries rearrangement, the
capacity of 70% HClO4 to catalyze the reaction of phenol acylation was investigated, since the only case of the use of this acid was the unsuccessful
attempt of Dorofeenko (CA 55, 24518i) to acylate phenol and resorcinol, which gave the esters instead of the ketones. Phenol (50 g.) was treated with
45 cc. Ac2O and 10 cc. 70% HClO4, the mixt. heated 30 min. on a water bath, left 24 h. at room temp., neutralized with 20% Na2CO3, 20 g. NaOH in 100
cc. H2O added, the aq. alk. soln. extd. with ether, then neutralized with 20% HCl, reextd. with ether, the ether evapd., and the product distd. to
give 51% 2-hydroxyacetophenone, b7 86, and the distn. residue dissolved in 20% NaOH, boiled 30 min., cooled, pptd. with 10% HCl, and recrystd. from
H2O with animal charcoal, to give 20.5% 4-hydroxyacetophenone, m. 108-9. Parallel Fries rearrangement of PhOAc under the same conditions gave the o-
and p-isomers in 38 and 19% yields, resp., thus confirming that the ester was formed at first and was then rearranged to the corresponding ketone.
Similar treatment of o-cresol (3 h. at 120) gave 55.5% 2-hydroxy-3-methylacetophenone, b10 105, 31% 3-methyl-4-hydroxyacetophenone. Treatment of
m-cresol (1 h. with no external heating, then 48 h. at room temp.) gave the o- and p-isomers, 62.8% 2-hydroxy-4-methyl- and 6%
4-hydroxy-2-methylacetophenone, b7 103, and m. 128% resp. Similar treatment of p-cresol (heating 1 h. at 100 then keeping 48 h. at room temp.) gave
53% 2-hydroxy-5-methylacetophenone, b7 101-3. Treatment of resorcinol (cooling, strong agitation, then 24 h. at room temp.) gave after neutralization
with Na2CO3 90% 2,4-dihydroxyacetophenone; similar treatment of resorcinol monoacetate gave 83% same product. Treatment of pyrocatechol (refluxing 3
h. on a water bath) gave 58% light-violet 2,3-dihydroxyacetophenone, m. 97.
Identical treatment of pyrogallol gave 44% 2,3,4-trihydroxyacetophenone, m. 173. Treatment of .alpha.-naphthol (1 h. at 115-20 on a sand bath) gave
after steam-distn. part of the 1-hydroxy-2-acetylnaphthalene formed, m. 100; further boiling of the resinous residue 1 h. in NaOH, neutralization, and
steam-distn. gave some further product, for a total yield of 40%. Salicylic acid was first dissolved in Ac2O-HClO4 at 100, the mixt. kept 0.5
h. at 120-30, cooled, H2O added, and the ppt. filtered off and recrystd. (H2O) to give 20-5% 3-carboxy-4-hydroxyacetophenone, m. 209-10,
phenylhydrazone m. 260; semicarbazone m. 222. Heating the reaction mixt. 1.5-2 h. at 120-30 and similar purifn. gave 71%
2-hydroxy-3-carboxyacetophenone, m. 148-9; phenylhydrazone m. 218; semicarbazone m. 240. The cause for the limited usefulness of HClO4 as a
catalyst in the Fries rearrangement was hence attributed to the oxidizing nature of this acid, which appeared only >170 for long durations in the
case of phenol acylation, but did not appear when the reaction temp. was kept <140 in the case of the Fries rearrangement. Its advantages over
other acid catalysts were: low amt. needed (0.1-0.2 mol per mol phenol compared to 1-2 mol AlCl3); no solvent needed as is the case with AlCl3 (the
use of solvents in the case of HClO4 did not improve the yields); and the preferential catalysis to the o-isomer, which in the case of some
phenols-pyrocatechol, pyrogallol, and salicylic acid-was the only isomer obtained, in contrast to the p-isomer obtained when AlCl3 was used, while in
the other cases the yields of o-isomer were greater than and the yields of p-isomer smaller than when AlCl3 was used as catalyst.
|
Whatever conditions are to be used, the Fries rearrangement of para-unsubstituted acyloxybenzenes rarely gives a clean ortho-rearrangement.
There are however a few examples of the Fries rearrangement of 5-substituted acylsalicylic acids in the literature, however these all have the
para-position in respect to the acyloxy group blocked (with Me, Et, F or I):
Inorganic Chemistry, 41 (2002) 3673-3683. (AlCl3)
J. Ind. Chem. Soc., 41 (1964) 833.
Journal of Medicinal Chemistry, 29 (1986) 538-549. (same method as ref above)
Journal of Chemical Research, Synopses (1985) 372-373. (AlCl3/MeNO2)
About the decarboxylation... The retro-Kolbe-Schmitt reaction procceds best the more hydroxy groups there are on the aromatic ring and the more
electron withdrawing groups there are para/ortho to these hydroxy groups. 3-Acetylsalicylic acid is barelly souitable, but might undergo such
decarboxylation under proper conditions. The literature is however more aboundant with Cu-catalysed decarboxylation of substituted salicylic acids
(eg, Helvetica Chimica Acta, 68 (1985) 945-948 where Cu2O/bipyridine/diglyme is used).
Also, making something as interesting as 3-acetylsalicylic acid just to decompose it to something as trivial and cheap as ortho-hydroxyacetophenone or
catechol is a bit dumb in my opinion. Both of these can be simply bought, as they are neither toxic or controled in any way, nor are they
prohibitively expensive. On the other hand 3-acetylsalicylic acid is not commercially available and its syntheses described in the literature are all
quite tedious.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
dean stark
Harmless
Posts: 12
Registered: 17-3-2011
Member Is Offline
Mood: No Mood
|
|
Thanks Nicodem.
The Fries rearrangement in the paper I mentioned seems to be selective to some degree depending on whether it is MWed or simply heated.
Now that I have the m.p. (from your quote), it should at least be possible to see if it works. 13 minutes in the microwave isn't too bad...
I will check out your suggestions for decarboxylation. Something's gotta work.
As for price and availability, my options are pretty limited for the time being.
|
|
|
adianadiadi
Harmless
Posts: 8
Registered: 22-2-2011
Member Is Offline
Mood: No Mood
|
|
Sorry for posting on an older post. However I would like to give a link showing Fries rearrangement of aspirin using AlCl3 as catalyst. I think it
works well.
http://www.angelfire.com/ny4/tecuci/synthesis.html
|
|
|
Random
International Hazard
Posts: 1018
Registered: 7-5-2010
Location: In ur closet
Member Is Offline
Mood: Energetic
|
|
Catechol can be prepared by the demethylation of guaiacol with aluminum chloride and hydriodic acid (from orgsyn.com)
Composition of a typical beech-tar creosote
Phenol C6H5OH 5.2%
o-cresol (CH3)C6H4(OH) 10.4%
m- and p-cresols (CH3)C6H4(OH) 11.6%
o-ethylphenol C6H4(C2H5)OH 3.6%
Guaiacol C6H4(OH)(OCH3) 25.0%
1,3,4-xylenol C6H3(CH3)2OH 2.0%
1,3,5-xylenol C6H3(CH3)2OH 1.0%
Various phenols C6H5OH— 6.2%
Creosol and homologs C6H3(CH3)(OH)(OCH3)— 35.0%
Creosote can be made from dry distillation of beech wood and the resulting sticky liquid can be used to isolate guiacol. About 25% of guiacol in beech
tar creosote.
Then guiacol can be demethylated as described above. I am currently experimenting with creosote and its compounds.
|
|
|
Dronami_inc
Harmless
Posts: 13
Registered: 10-2-2011
Member Is Offline
Mood: No Mood
|
|
I think that very interesting way would be replacing the methyl groups in o-xylene with OH-group. Certainly not in one step. More interesting in
theory, how to implement it.
 ---> --->

IMHO, the first phase - the oxidation to phthalic anhydride or acid
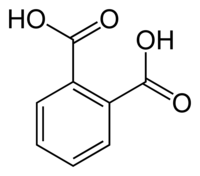
|
|
|
paw_20
Harmless
Posts: 32
Registered: 14-8-2012
Location: United States
Member Is Offline
Mood: Curious
|
|
hot KMnO4 and then reduction with LAH, not much else reduces carboxylic acids. But it isn't easy to separate o-xylene from it's isomers.
|
|
|
phendrol
Harmless
Posts: 34
Registered: 13-6-2012
Member Is Offline
Mood: sock puppet
|
|
Catechol from salicylaldehyde
I want to try the preparation of catechol from salicylaldehyde.
| Quote: |
To a solution of 122 g. (1 mole) of pure salicylaldehyde (Note 1) in 1000 cc. of normal sodium hydroxide solution at room temperature, is added 1420
g. (1.2 moles) of 3 per cent hydrogen peroxide. The mixture darkens slightly in color and the temperature rises to 45–50°. The solution is allowed
to stand for fifteen to twenty hours, whereupon a few drops of acetic acid are added in order to neutralize any excess alkali, and the solution
evaporated to complete dryness on the water bath under reduced pressure. The solid residue is finely crushed and warmed nearly to boiling with 500 cc.
of toluene; the mixture is then poured into the folded filter paper of an extraction apparatus (p. 375) and extracted with boiling toluene for five
hours. The toluene is allowed to cool and is decanted from the catechol, which crystallizes out. The insoluble material is again ground up and
extracted in the apparatus with the decanted toluene. |
Organic Syntheses, Coll. Vol. 1, p.149 (1941); Vol. 3, p.28 (1923).
The only thing that makes me hesitate is that I don't have a vacuum pump, so I can't evaporate all the water under reduced pressure. I'm afraid that
simply boiling of the water under atmospheric pressure will cause loses because a part of the catechol will sublime.
Any thoughts on that anyone? 
|
|
|
hyfalcon
International Hazard
Posts: 1003
Registered: 29-3-2012
Member Is Offline
Mood: No Mood
|
|
Here's the live link to your paper.
http://www.google.com/url?sa=t&rct=j&q=&esrc=s&a...
Any reason you can't evaporate at a lower and slower temperature? Possibly over a desiccant?
[Edited on 19-10-2013 by hyfalcon]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
what about making catechol by oxidising ortho nitrophenol with DMDO or peroxide ?
ortho nitrophenol can be selectively synthesised using clay +nitrating reagent
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
Are you still trying to apply Meisenheimer complex chemistry to non-adduct nitrobenzenes? Because that won't work. Unless you have a reference
indicating conditions where this has been confirmed, it is important to realize that substituents such as those withdrawing electrons in a specific
manner in Meisenheimer complexes can alter reactions and stability in much the same way the Jahn-Teller effect explains anti-aromatic reactivity.
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
one of the resonance structures of para nitrophenol has a similar structure like that meisenhiemer complex,so I wondered maybe ortho nitro phenol
could also exist in that form,because nicodem had said that ortho and para amino phenol had similar quinoid structures.

|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
Interesting idea, but I see quite a distinction between a minor resonance contributor (see the unfavorable charge separation on the nitrogen and both
oxygens?) with a stable (isolated even) adduct.
Edit: Also, do note you technically did not draw a resonance structure as one figure is protonated at the phenol and the other assumes deprotonation.
Resonance structures are between either structures of the acid itself with arrow pushing, or of the phenoxide with arrow pushing, but not between an
acid and its conjugate base, which require an equilibrium arrow by convention rather than resonance notation. Consider where the cation or solvation
shells would best fit.
[Edited on 16-5-2015 by Chemosynthesis]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
CuReUS, i think you are right about resonance structure (althoug you will need a base to obtain it), but I see no way to substitute the nitro group,
because you can't do Nef reaction on nitroalkene - you need to reduce it first. Maybe you see some other known mechanism that will allow you to
substitute the nitro group?
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
I realised later that there was a huge flaw in my idea.Even if nef reaction could be done on the nitronate,it would not tautomerise to a phenol as
there is no Hydrogen
|
|
|
clearly_not_atara
International Hazard
Posts: 2694
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Other idea:
1. Esterify acetylsalicylic acid with -anything- to get, say, O-acetylsalicylic acid ethyl ester. Use some sort of non-nucleophilic base or
dehydrating conditions so you don't cleave the acetate.
2. Use NaNH2/LiNH2/NaOiPr to catalyze an intramolecular Claisen condensation: the acetyl group deprotonates and is attacked by the ortho- carboxyl
group to give 2,4-dioxobenzopyran. Sodium/lithium amide should work fine here; it's not as nucleophilic as it looks. KOH/DMSO might work, actually.
Aprotic solvents are ideal.
3. A Dakin reaction produces catechol and malonic acid. Malonic acid can be extracted from the rxn by precipitation with Ca2+ since it's
kind of interesting too. If you're clever and lucky and perform the rxn in ethanol you could generate diethyl malonate directly.
[Edited on 24-5-2015 by clearly_not_atara]
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by CuReUS  | | I realised later that there was a huge flaw in my idea.Even if nef reaction could be done on the nitronate,it would not tautomerise to a phenol as
there is no Hydrogen |
Not a tautomer if it's short a hydrogen.
|
|
|
Nicodem
|
Thread Split
25-5-2015 at 07:04 |
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
I tried this synthesis using the 5.1g of salicylaldehyde that I had recently synthesized.
Using a scale factor of 0.05 I added 3% H202 and 1M NaOH to a 250mL beaker. Then I added the salicylaldehyde and stirred with a magnetic stirrer.
The solution went from yellow to dark red. Temperature rose to 50°C as indicated in the OrgSyn procedure.
After setting overnight I added a few drops of acetic acid to bring to neutral. I overshot a bit resulting in a pH of 6.
I placed the beaker in a water bath and heated it all day long. Then I changed to a silicone bath controlled to 100°C. Finally it dried to a dark,
viscous tar having the smell of phenol..
Today I added 28 ml of toluene, warmed to near boiling, and stirred. There was no color change of the toluene, indicating to me, no dissolution.
This synthesis appears to be a failure. Most likely what I have made is just a tar composed of polymerized catechol.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Wooohoo !
I am sooo happy that my own tarry products are not totally abnormal.
I thought i was just stupid.
|
|
|
Pumukli
National Hazard
Posts: 686
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Magpie |
Today I added 28 ml of toluene, warmed to near boiling, and stirred. There was no color change of the toluene, indicating to me, no dissolution.
This synthesis appears to be a failure. Most likely what I have made is just a tar composed of polymerized catechol.
|
Hey, Magpie, maybe you are just one step away from success!
The original article says that the solid residue must be extracted with toluene, then repeatedle extracted again, for several hours, etc.
So you got to the solid residue (although tarry) and extracted it with toluene.
Then you complain about "no color change". Why?
I think catechol is colorless, at least it should be!
Then you wrote what you extracted smelled phenol-like. Woo-hoo! Isn't catechol expected to have at least a faint phenol-like odour? :-)
Why not evaporate (distill off) the toluene to see if you really got something dissolved in it? Maybe you got it, even if the yield is probably not
astronomical, but I would not care less at this stage. You made a nice (and tedious) salicylaldehyde synthesis, please don't let it go just like this!
I'm curious about the final step - and I'm fairly sure some SM members feel the same!
[Edited on 26-6-2015 by Pumukli]
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Pumukli, I agree. I just lost patience when I saw the tar. I let it sit outside until the toluene evaporated. Then this morning I removed most of
the tar with a spatula. Then I added some NaOH and a little water just to clean out the beaker.
[Edited on 27-6-2015 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
stoichiometric_steve
National Hazard
Posts: 819
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
i have some catechol for sale, if that's what you're after?
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
| Quote: Originally posted by Magpie |
This synthesis appears to be a failure. Most likely what I have made is just a tar composed of polymerized catechol.
|
Here's what the product looked like. I meant to post this in the original post but forgot.

The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
That NaOH should be worked on. Perhaps the toluene was unable to access anything it could dissolve.
|
|
|
zed
International Hazard
Posts: 2277
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Lots of faked results out there. But, Organic Synthesis procedures....Well, thus far, every one I have tried has worked.
|
|
|
| Pages:
1
2
3
4
5 |









