| Pages:
1
2 |
garage chemist
chemical wizard

Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
Preparation of trinitromethane
I think that this compound is worth a seperate thread.
The method has been briefly discussed before, and this
patent proved to be most useful (scroll down, it gets interesting).
The biggest problem lies in extraction of the nitroform. Because only 9,8g of nitroform are formed from 140ml of 98% nitric acid, the recycling of the
acid is absolutely neccessary, too.
Some patents on the extraction are mentioned, like these:
no.1no.2
However, the pictures are somehow messed up.
Does anyone know how to seperate a mixture comprised of lots of nitric acid and a little nitroform?
[Edited on 20-12-2004 by garage chemist]
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Wouldn't it be easier (less wasteful) to react iodoform HCI3 with Ag/NaNO2 in DMF/ether or whatever, according to reactions described in the nitroalkane thread? I remember that iodoform is made quite easily, so the only real trouble would be to get the solvents & the silver nitrite.
On the note of extraction - other than distillation, I don't see a method with which you can recycle the HNO3. It sounds unlikely that you can
recycle it properly anyway, as you lose a lot of acid as NO2.
Anyway, trinitromethane is a fairly strong acid, as far as I remember. So if you neutralise those 140 ml, get rid of the NaNO3/whatever, you should
be left with the salt of the nitroform - which, aside from this, has probably a different colour/solubilities than KNO3. Alternatively, you could just
keep the mix boiling until you got rid of ALL HNO3, beign left with nitroform. A formidable waste though.
PS Could you please put those links into HTML tags? This page is about 1 foot wide  Also, those links don't seem to work in the first place. Also, those links don't seem to work in the first place.
[Edited on 20-12-2004 by chemoleo]
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
I also thought of CHI3 + NaNO2, but does NaNO2 work? Silver nitrite would of course give a better yield (forming insoluble AgI) but it would be
difficult and expensive to get.
It might also be hard to find a solvent in which both CHI3 and NaNO2 are soluble. You said ether/DMF?
This might be worth an experiment, as I have about one gram of CHI3 with no use for it.
I have ether, but no DMF. I could get DMF though if I really needed it.
|
|
|
Esplosivo
Hazard to Others
Posts: 491
Registered: 7-2-2004
Location: Mediterranean
Member Is Offline
Mood: Quantized
|
|
According to this MSDS Iodoform is said to be soluble in alcohol. NaNO2 is usually dissolved in propanone which isn't hard to get either. Hope this helps.
Theory guides, experiment decides.
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
That's good, NaNO2 is also soluble in ethanol (not much, but likely enough to make the reaction possible).
Now, the reaction conditions. The MSDS from jtbaker lists neither silver nitrite nor sodium nitrite as incompatibilities, so the reaction most likely
proceeds slow and under heat.
Do you think I should try and see what happens when I mix CHI3 and NaNO2 in Ethanol and heat it?
|
|
|
Esplosivo
Hazard to Others
Posts: 491
Registered: 7-2-2004
Location: Mediterranean
Member Is Offline
Mood: Quantized
|
|
Well usually such reactions do not involve the use of much heat, warming is generally carried out. This I've done with monohalidealkanes though,
and not with trihalidealkanes. You could try out a rxn between small amounts of reagents on the qualitative scale as a test. If the yellow colouration
of the CHI3 disappears than the rxn must have suceeded.
Theory guides, experiment decides.
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
Before I try out the CHI3 + NaNO2 reaction:
How toxic is trinitromethane? Is it carcinogenic?
And at last, what is its boiling point? Can it be safely distilled or do I need to use another way of seperating it from the ethanol?
Sorry for asking this here, but I found nothing useful with google.
|
|
|
Nick F
Hazard to Others
Posts: 439
Registered: 7-9-2002
Member Is Offline
Mood: No Mood
|
|
I'm not too sure about haloforms and nitrites. Firstly, if it was that easy then no-one would want to use huge excesses of nitric acid on
isopropanol or ketene or acetylene or any of those other methods. They're not very economical, and all of them involve oxidation, which means
lots of NOx to deal with.
Secondly, if you draw a space-filling model of a haloform roughly to scale you will see that they are VERY sterically hindered. Fluoroform
wouldn't be too bad, but F- is a pretty rubbish leaving group. Leaving ability increases from Cl to I, but so does size and therefore hinderence.
Maybe if you used AgNO2 though, any halide which left would be instantly mopped up and multiple SN1 reactions could then be forced to give your TNM.
But you will likely have difficulties with isomerism - each halogen could be replaced by a nitro group or a nitrite ester group...
I don't know, maybe it would work, I just don't get a good feeling about it!
I think the best route would be an oxidative nitration of something with a good method for acid recovery. Probably some sort of two-phase extraction
could be used, but your solvent had better be inert (conc. strong acids, explosive oxidisers...). Chloroform perhaps? You might not find a solvent
which dissolves on and none of the other, but I bet you could increase the nitroform:nitric acid ratio using a solvent extraction and process that
further with base and fractional crystalisation. And it would stop you from having to distil such a compound. I wouldn't distil it, I'd be
too scared.
I don't like the idea of heating it with ethanol, either. In fact I don't like the idea of nitroform going anywhere near ethanol!
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Do you realise that tetranitromethane can be made from iodopicrin I3CNO2 and AgNO2? There is a reference regarding this in the OrgSyn
tetranitromethane prep from acetic anhydride and HNO3 (which I'd love to get my hands on).
I don't see why this is radically different to making trinitromethane from HCI3 and AgNO2.
Long time no see Nick!
[Edited on 21-12-2004 by chemoleo]
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
@ Nick: You're right, a mixture of ethanol and TNM is supposedly explosive. Another thing to care about.
AgNO2 could be made by bubbling N2O3 through a suspension of Ag2O.
But I'll try the synthesis out first with NaNO2.
Here
is the preparation of Tetranitromethane from orgsyn, with the reference chemoleo mentioned.
I'd also like to see the Tetranitromethane prep from iodopicrin and AgNO2, one could just use iodoform instead of iodopicrin and it would likely
give nitroform.
How toxic is Trinitromethane? I won't try its synthesis before I have information on this.
|
|
|
Nick F
Hazard to Others
Posts: 439
Registered: 7-9-2002
Member Is Offline
Mood: No Mood
|
|
Good to be back chemoleo! I don't do much online stuff while away at Uni...
Anyway, that's very interesting about iodopicrin -> TeNM. I wonder if iodopicrin was used because of its reactivity, or just because it's
less volatile and hence less irritating than the cheaper chloropicrin?
Iodoform is super-easy to make, and should react OK I think. The C will be less electrophilic at first, but also more vulnerable due to the little H
compared to the huge NO2 on iodopicrin. It is certainly worth trying!
TNM ain't that toxic, just don't go around eating it! (or breathing it, or touching it!)
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Experiment!
Well, this was put to the test.
Iodoform (a few spatula tips), made according to the method here, was dissolved in ether (25ml), which formed a pale yellow solution.
To this AgNO2 powder (yellow grey) was added, also a few spatural tips, keeping the solution on ice all the while. The AgNO2 didn't dissolve.
Remarkably, the solution did turn a dark yellow after an hour or so, while the AgNO2 powder that was previously dense and settled at the bottom became
fluffy, and a dark grey colour, with blacker patches.
When adding a couple of ml of this to H2o, the yellow colour remained in the etheric fraction  , which is unfortunate because the TNMe should really dissolve in H2O. No visible reaction of the etheric extract with 34% formaldehyde
either - but again part of the problem could be because the solutions don't mix. , which is unfortunate because the TNMe should really dissolve in H2O. No visible reaction of the etheric extract with 34% formaldehyde
either - but again part of the problem could be because the solutions don't mix.
Anyway, it definitely looks encouraging due to the colour change etc. I shall add some more AgNO2, to see what happens with it. Lateron I might try
the same wtih NaNO2, and ethanol.
I am not particularly worried about det. hazard etc becuase the amounts used are minute compared to the large solvent excess.
[Edited on 22-12-2004 by chemoleo]
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Hmm, I am not so sure whats going on anymore.
Just added some of the dark yellow solution to fresh AgNO2, and the solution turned a light yellow once again?!?!? - i.e. pretty much identical to
what it was when I started.
Very peculiar. I guess next time I should make sure that light is excluded.
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
Esplosivo
Hazard to Others
Posts: 491
Registered: 7-2-2004
Location: Mediterranean
Member Is Offline
Mood: Quantized
|
|
Note that the reaction should produce a precipitate of silver iodide, which has a yellowish colour. This might be causing the yellow colour in your
case, but I'm not sure.
Theory guides, experiment decides.
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
There is a patent involving the preparation of nitroform from acetylene and HNO3. Do a search on the European patent web site. As far as I know,
nitroform and tetranitromethane are supposed to be nasty toxic and carcinogenic.
|
|
|
Madandcrazy
Hazard to Others
Posts: 117
Registered: 11-5-2005
Member Is Offline
Mood: annoyed
|
|
Rather the trinitromethane is prepared over dinitromethane from nitromethane, temperature and concentration of the acid is pregnant. Maybe useful for
substitution
when chloronitromethane or chlorodinitromethane is reacted with methylenediamin, and aluminium carbonate to
CH2(NHCH2NO2)2 or CH2(NHCH[NO2]2)2.
[Edited on 9-2-2006 by Madandcrazy]
|
|
|
Endo
Hazard to Others
Posts: 124
Registered: 5-1-2006
Location: USA
Member Is Offline
Mood: Cold
|
|
| Quote: | Originally posted by chemoleo
Do you realise that tetranitromethane can be made from iodopicrin I3CNO2 and AgNO2? There is a reference regarding this in the OrgSyn
tetranitromethane prep from acetic anhydride and HNO3 (which I'd love to get my hands on).
|
In the book Chemistry of powder and Explosives by Davis, on page 126 he discusses the formation of tetranitromethane by refluxing TNT or picric acid
with nitric acid, then distilling off the tetranitro methane.
|
|
|
Hoveland
Harmless
Posts: 20
Registered: 20-7-2010
Member Is Offline
Mood: No Mood
|
|
Trinitromethane (nitroform, the acid of nitroformate) can probably be easily prepared through an uncommon pathway. DiNitroso, monoNitro methane can be
prepared from nitroso methane and N2O4 (or presumably NO2)
B G Gowenlock and I Batt
'The isomerisation of nitrosomethane to formaldoxime', Theochem-Journal of Molecular Structure, 454 1998: 103-4.
B G Gowenlock, B King, J Pfab and M Witanowski
'Kinetic studies of the reaction of some nitrosoalkanes with nitrogen dioxide', Journal of the Chemical Society, Perkin Transactions 2, 1998: 483-5.
In my opinion, excess treatment with nitrogen dioxide would result in trinitromethane, as NO2 oxidizes nitroso.
"The oxidation of nitrosobenzene by nitrogen dioxide in carbon tetrachloride has been re-examined"
" There is an early report 5 that a small quantity of nitrobenzene was formed when dry chloroform solutions of nitrogen dioxide and nitrosobenzene
were allowed to stand at 22 8C for 39 h."
J. Chem. Soc., Perkin Trans. 2, 1997, 1793 - 1798, DOI: 10.1039/a700258k
Kinetics of the oxidation of aromatic C-nitroso compounds by nitrogen dioxide
Brian G. Gowenlock, Josef Pfab and Victor M. Young
"Hydroxylamine, an intermediate in ammonia oxidation, reacts with formaldehyde to form formaldoxime" Methanol and Formaldehyde Oxidation by an
Autotrophic Nitrifying
Bacterium by PA Voysey - 1987
|
|
|
dunmail
Harmless
Posts: 6
Registered: 16-7-2007
Location: UK
Member Is Offline
Mood: damp
|
|
I wonder would there be any benefit in having alkine conditions and trying to extract the nitroform as a nitroformate of e.g. sodium??
[Edited on 30-8-2010 by dunmail]
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
how hydrogen peroxide reduces tetranitromethane
It is well known that tetranitromethane can be reduced to nitroformate salts using an alkaline solution of hydrogen peroxide. This is the most usual
route for preparing trinitromethane.
Here is the procedure from RS:
Prepare a solution of 168 g of potassium hydroxide in 350 mL of water in a round-bottomed 1000-mL Florence flask, and cool to 5 °C with a salt-ice
bath. While stirring, add 108 mL of 30% hydrogen peroxide to the solution. Next, add 117 mL of tetranitromethane at a rate which keeps the temperature
at 20-25 °C, add while stirring. The temperature is then allowed to rise to 30 °C over 15 minutes. The bright yellow solid, that should have formed,
is filtered to collect it using glass filter paper because of its high acidity, washed with anhydrous methyl alcohol, then anhydrous ethyl ether, and
finally air dried to give 100% of the potassium salt of trinitromethane. The salt is suspended in anhydrous ethyl ether and anhydrous hydrogen
chloride gas is passed in until the yellow color disappears. The white precipitate of potassium chloride is filtered off and washed with anhydrous
ethyl ether. The ethyl ether is evaporated from the filtrate and additional washings at reduced pressure give 85-90% of crude trinitromethane which
can be purified by sublimation.
Although usually an oxidizer, in some reactions hydrogen peroxide can act as a reducing agent. For example, it reacts with hypochlorite to form
chloride and oxygen gas. Similarly, an alkaline solution of H2O2 reduces Cl2 to Cl- ions.
I wrote out a possible reaction mechanism below.
[Edited on 5-5-2011 by AndersHoveland]
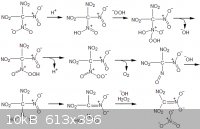
Trinitroacetonitrile
Hantzsch and Rinckenberger obtained the ammonium salt of trinitromethane by treating tetranitromethane with aqueous ammonia.
Most salts of trinitromethane derive from the aci-form. However, the silver and mercuric salts exist in two forms: colourless and yellow. This may
indicate that two forms of these salts - nitro and aci - can exist.
Trinitroacetonitrile
Trinitroacetonitrile can be synthesized by the nitration of cyanoacetic acid with a solution of sulfur dioxide and 98+% concentrated nitric acid in
carbon tetrachloride, with 73-77% yields. The trinitroacetonitrile can be stored as a solution in the carbon tetrachloride, and need not be isolated
for further use on other reactions.
NCCH2C(=O)OH + (3) HNO3 + (3)SO2 -- > NCC(NO2)3 + CO2 + (3) H2SO4
Trinitroacetonitrile is a colorless, camphor-like, crystalline compound melting at 41.5 °, and detonating violently at 220°. It hydrolyzes to carbon
dioxide and the ammonium compound of nitroform by water or alcohol at ordinary temperatures.
"Nitroacetonitrile has been prepared by treating methazonic acid with thionyl chloride SOCl2 in ether. That the compound so obtained is
nitroacetonitrile follows from the fact that it yields a-nitroethenylamino-oxime with hydroxylamine, and gives the nitrolic acid reaction. Its
formation from methazonic acid proves the correctness of the formula. (methazonic acid has the formula HON=NCHCH2NO2, and is well discussed elsewhere
on this forum). Nitroacetone, NCCH2NO2 ,is obtained as a fairly stable yellow oily liquid. Wen pure, it may be distilled under reduced pressure
(boiling point 96 ° under 14mmHg reduced pressure). It does not seem to be explosive, neither is the ammonium salt, which crystallizes in slender,
yellowish-white needles, decomposing at 130-135°. The silver salt, obtained as a brown precipitate, however, is a sensitive explosive.
A-Nitroethenylamino-oxime (NO2)CH2C(NH2)=NOH, obtained by the action of hydroxylamine on nitroacetonitrile, forms yellow crystals and decomposes
suddenly at 108°."
Journal of the Royal Society of Chemistry (Great Britain), Volume 94. p.327 (year 1908)
It is known that dinitroacetonitrile can be nitrated to trinitroacetonitrile, so nitroacetonitrile probably can be similarly nitrated.
Shishkov (and later Steiner) claimed to have obtained trinitroacetonitrile by treating the sodium salt of fulminuric acid with a mixture of nitric and
sulphuric acid, but it was later shown that the compound which is obtained from this reaction is not identical to trinitroacetonitrile.
“Nitroform (Trinitromethane), CH(N03)3, is obtained in the form of its ammonium salt by the decomposition of trinitroacetonitrile with water.” (L.
Schischkoff, Ann., 1857, 10 3, p. 364).
Direct nitration of acetonitrile?
I am unsure, but I think it may be possible that trinitroacetonitrile could be prepared by nitration of acetonitrile using nitronium
tetrafluoroborate. Although acetonitrile is commonly used a solvent for the nitration of other reagents by nitronium tetrafluoroborate, apparently
without significant reaction of the acetonitrile, it may likely be that the acetonitrile does in fact slowly react, but much less rapidly than the
nitration of the other reagent being nitrated. There is a reaction, which was developed by Olah, in which alkanes, which are generally fairly inert at
room temperature, can be nitrated using nitronium salts (procedure below). Thus it seems probably that acetonitrile could be similarly nitrated.
Procedure for the Nitration of Alkanes using Nitronium Hexafluorophosphate:
Dichloromethane and nitroethane were dried by refluxing over calcium hydride followed by distillation. 1,1,2-Trichlorotrifluoroethane (Freon-113 was
dried over phosphorus pentoxide under dry nitrogen and then distilled. Trifluoromethanesulfonic acid (triflic acid) was freshly distilled under dry
nitrogen before use. Nitronium hexafluorophosphate was prepared from fuming nitric acid, anhydrous HF, HPF6 (60%), and PCl5 following a recently
developed procedure (G.K.S.P., D.K. Padma, P.R., D. Adamson, and G.A.O., unpublished results).
Nitronium hexafluorophosphate (3.82 g, 20 mmol) was added under dry nitrogen into a flame-dried 50-ml reaction flask equipped with a magnetic stirrer,
condenser, nitrogen, and alkane inlet. Dry dichloromethane (20 ml) then was added, and the appropriate gaseous alkane was passed into the suspension
at ambient temperature with rapid stirring. The reaction mixture was monitored by analysis of periodically removed samples. Introduction of the alkane
was stopped until the yield of nitroalkane did not increase further. The reaction mixture then was quenched with water and extracted with
dichloromethane. The organic layer was washed successively with 5% aqueous NaHCO3 solution and water, dried over MgSO4. After evaporation of the
solvent, the nitro product yields were found to be based on the amount of NO2+PF6− used.
NO2( +) PF6( − ) is much preferred over NO2( + ) BF4( − ) as a nitrating agent because it is substantially more soluble in dichloromethane
(about 4 mol%), the latter is practically insoluble in dichloromethane. In nitroethane the solubility of NO2+PF6− is more than 25 mol%.
Information about the preparation of Nitronium Tetrafluoroborate NO2( + ) BF4( - ), can be found at my site:
http://sites.google.com/site/ecpreparation/nitronium-tetrafl...
Salts of Dinitromethane
Dinitromethane forms salts which are generally unstable and decompose at temperatures not very much above 100°C.
[Edited on 6-5-2011 by AndersHoveland]
_______________________
PLEASE DON'T DOUBLE POST
[Edited on 6-5-2011 by quicksilver]
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
Preparation of Nitroform from Nitric Acid and Isopropanol:
A 250 ml three-necked flask was fitted with a mechanical stirrer, a thermometer and a dropping funnel. 140 ml (3.33 moles) of 98% nitric acid was
introduced into the flask. The acid was warmed to about 60.degree. C. and 20 ml (0.26 mole) of isopropyl alcohol was added dropwise over a 10-minute
interval. External cooling was used to maintain the temperature at 60.degree. C. The solution was then heated to a temperature of about 70.degree. C.
and held at this temperature for 2 hours. Substantial quantities of brown gaseous fumes evolved during this nitration. The solution subsequently was
cooled to ambient temperature and analyzed for nitroform content. The yield of nitroform was determined to be 9.8 gm (approximately a 25% yield).
To obtain significant yields of the desired trinitromethane it is essential that the isopropyl alcohol be introduced into an excess of nitric acid.
Thus, the molar ratio of nitric acid to isopropyl alcohol will be in excess of about 8:1. Too great an excess of nitric acid will, of course, increase
the cost of the method, and will require an unnecessary amount of nitric acid to be distilled and recycled to the process. Thus, the molar ratio of
nitric acid to isopropyl alcohol generally is maintained within a range of from about 10 to 25, and preferably within a range of from about 15:1 to
20:1.
The reaction temperature is not particularly critical, provided, of course, that the temperature must be sufficiently high to maintain the mixture of
reactants in a liquid phase. In addition, the temperature should not be too high, otherwise substantial gas evolution takes place with little or no
formation of nitroform. Therefore, the temperature generally has been maintained within a range of from about 25.degree. to 85.degree. C. and
preferably within a range of from about 40.degree. to 70.degree. C. The time required for the reaction will vary with temperature, pressure ratio of
reactants, etc. Generally, a time of from about 1 to 5 hours is sufficient to react substantially all of the isopropyl alcohol to form the desired
trinitromethane.
Yields of up to 50-58% have been obtained from a modification of this procedure.
|
|
|
Dr.Q
Harmless
Posts: 24
Registered: 18-11-2012
Member Is Offline
Mood: No Mood
|
|
| Quote: |
Preparation of Nitroform from Nitric Acid and Isopropanol: A 250 ml three-necked flask was fitted with a mechanical stirrer, a thermometer and a
dropping funnel. 140 ml (3.33 moles) of 98% nitric acid was introduced into the flask. The acid was warmed to about 60.degree. C. and 20 ml (0.26
mole) of isopropyl alcohol was added dropwise over a 10-minute interval. External cooling was used to maintain the temperature at 60.degree. C. The
solution was then heated to a temperature of about 70.degree. C. and held at this temperature for 2 hours. Substantial quantities of brown gaseous
fumes evolved during this nitration. The solution subsequently was cooled to ambient temperature and analyzed for nitroform content. The yield of
nitroform was determined to be 9.8 gm (approximately a 25% yield). To obtain significant yields of the desired trinitromethane it is essential that
the isopropyl alcohol be introduced into an excess of nitric acid. Thus, the molar ratio of nitric acid to isopropyl alcohol will be in excess of
about 8:1. Too great an excess of nitric acid will, of course, increase the cost of the method, and will require an unnecessary amount of nitric acid
to be distilled and recycled to the process. Thus, the molar ratio of nitric acid to isopropyl alcohol generally is maintained within a range of from
about 10 to 25, and preferably within a range of from about 15:1 to 20:1. The reaction temperature is not particularly critical, provided, of course,
that the temperature must be sufficiently high to maintain the mixture of reactants in a liquid phase. In addition, the temperature should not be too
high, otherwise substantial gas evolution takes place with little or no formation of nitroform. Therefore, the temperature generally has been
maintained within a range of from about 25.degree. to 85.degree. C. and preferably within a range of from about 40.degree. to 70.degree. C. The time
required for the reaction will vary with temperature, pressure ratio of reactants, etc. Generally, a time of from about 1 to 5 hours is sufficient to
react substantially all of the isopropyl alcohol to form the desired trinitromethane. Yields of up to 50-58% have been obtained from a modification of
this procedure.
|
I dont get it . I mean how is it possible synthesis nitroform from isopropanol. Can anyone wirght the mechanism ?
Shouldn't iso-propyl nitrate form ?
http://www.sciencemadness.org/talk/viewthread.php?tid=14492
[Edited on 7-12-2012 by Myeou]
|
|
|
starman
Hazard to Others
Posts: 318
Registered: 5-7-2008
Location: Western Australia
Member Is Offline
Mood: No Mood
|
|
The admin gifted this member with the epithet " Do Not Make the Mistake of Believing What I Say " for a reason!
Chemistry- The journey from the end of physics to the beginning of life.(starman)
|
|
|
Dr.Q
Harmless
Posts: 24
Registered: 18-11-2012
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by starman  | | The admin gifted this member with the epithet " Do Not Make the Mistake of Believing What I Say " for a reason! |
no no ... i mean seriously , i read a few synthesis method like acetylene through nitric acid , acetic anhydride and nitric acid .
But none of them comes logical . Can someone wright the mechanism of them. I mean that methods are really work ?
[Edited on 8-12-2012 by Myeou]
|
|
|
killswitch
Hazard to Others
Posts: 209
Registered: 8-7-2011
Location: is a relative concept
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by starman | | The admin gifted this member with the epithet " Do Not Make the Mistake of Believing What I Say " for a reason! |
His a lot of his shit works, dude. Take the bad with the good. I got 2 milligrams of trinitroazetidine using the procedure on his site. He was
considerate enough to warn of the low yield in advance, so I wasn't that disappointed.
I don't have NMR or IR access at the moment, but it gave off a very distinctive BANG when subjected to my jerry-rigged mini doom laser made out of two
cannibalized blue ray players and some fiber optic cable. None of the intermediates in his process detonate like that, so I'm inclined to give him the
benefit of the doubt.
Of course, he posts new things faster than anyone could hope to verify, so treat him like Wikipedia: a helpful source for helpful sources.
|
|
|
| Pages:
1
2 |










