| Pages:
1
2
3
4 |
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: | Originally posted by Vitus_Verdegast
In my experience when H2SO4 is used no sulfate precipitates, I don't think it would precipitate from a concentrated acetic acid/alcohol mixture.
When I used a mixture of 80% acetic acid, ethanol and conc. H2SO4, there was a more profound ethyl acetate smell than when 29% HCl was used, this
might increase the solubility of the P2NP somewhat.
Although in practice this doesn't seem to matter much, and I also prefer to use HCl. |
The sulfate of phenylisopropylamine , benzedrine sulfate has a very low almost nil solubility in alcohol and I would expect it to precipitate in the
electrolyte where it would be present in substantial amount unless there was a whole lot of water present . Maybe enough acetic acid would change
that but I doubt it . You must have had a pretty thin batch going there if it didn't just set up solid 
[Edited on 5-8-2006 by Rosco Bodine]
|
|
|
Vitus_Verdegast
Hazard to Others

Posts: 297
Registered: 5-12-2004
Location: Ottoman Empire
Member Is Offline
Mood: tea time
|
|
Of course, since I do not have the required license, I did not try it on unsubstituted P2NP.
I did try this for several substituted phenethylamines and amphetamines, every time at a 5-10g scale per +- 200ml solvent, never had any problems.
[Edited on 5-8-2006 by Vitus_Verdegast]
Sic transit gloria mundi
|
|
|
d0c
Harmless
Posts: 9
Registered: 4-8-2006
Member Is Offline
Mood: No Mood
|
|
Well boys and girls,
this would lead us to another path that could be explored.
If one uses anhydrous Ethanol Solution with H2SO4 acid and the sulfate really precipitates (what has already been observed by SWIM, but not analysed)
this would make the technique of purification even easier.
It would be possible to decant the solution and clean the precipitated substrate with nonsolving solvents and one would have the final product in 1
cleaning step instead of afterwards acid/base procedure.
could one describe what the precipitated amine looks like?
SWIM has observed yellow clumps near the cathode is it IT?
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
No shortcuts are recommended in the purification .
If you are not going to perform the usual isolation steps
including vacuum fractionation of the freebase , then
you really shouldn't be making this stuff in the first place .
The crude freebase is a brown oil which has the distinctive heavy aromatic odiferous
" amine stench "
and the vapors themselves are so physiologically active
that a good whiff is about like getting yourself plugged into a light socket a minute or so after inhalation you will be " wide awake " ....if you catch my meaning ,( sort of like the Mazda commercial Zoom Zoom Zoom ) ,
so you do not want to be exposed to the vapors during manipulations . And the stuff is highly basic and will form a carbonate on long exposure to the
air from the CO2 in the air . After vacuum distillation , the freebase is a nearly clear oil , perhaps with a pale brown tint to it like cooking oil ,
but almost colorless . The methylenedioxy variant tends towards a slight pink color .
[Edited on 5-8-2006 by Rosco Bodine]
|
|
|
d0c
Harmless
Posts: 9
Registered: 4-8-2006
Member Is Offline
Mood: No Mood
|
|
Hello,
SWIM did another experiment on saturday. Since no car battery was available so quickly a setup as described with Cu cathode and HCl acidified
elektrolytes was used with a small amount of P2NP. 12-14,5V @ 1.7-2.3Aand 45°C in a flower pot.
SWIM analysed what could structually happen and since the color change to cherry red happens quiet rapidly SWIM assume the reason is a polymerization
reaction after reduction step 1 at the cathode. Something seems to inhibit further reduction. After evaporation there is a RED TAR.
There could be 3 reasons:
1. cathode material should be Pb.
2. 1.7-2.3A is not enough, but the flower pot doesn't let more through. Does anyone know how to increase permeability of the flower pot?
3. The voltage was too high, as SWIM asked in his posts before what is the perfect voltage (aprox.)?
SWIM knows it depends on the H2 generation, but on the other side there is a difference if I use 1.3V or 13V...
Has anyone an idea?
One more thing. It needs to be verified the P2NP is P2NP, else all experimentation is useless. Does anyone have a chemical reduction method (except
with Hg salts!) to reduce the P2NP in order to verify the electrochemical experiments are usefull e.g. with NaBH4, PtO2 or similar pathes that atleast
work, compared to Urushibara...
Thank you very much for support.
SWIM promises once it works to write a large, detailed PDF with all aspects, theory, practice example and much more.
[Edited on 6-8-2006 by d0c]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
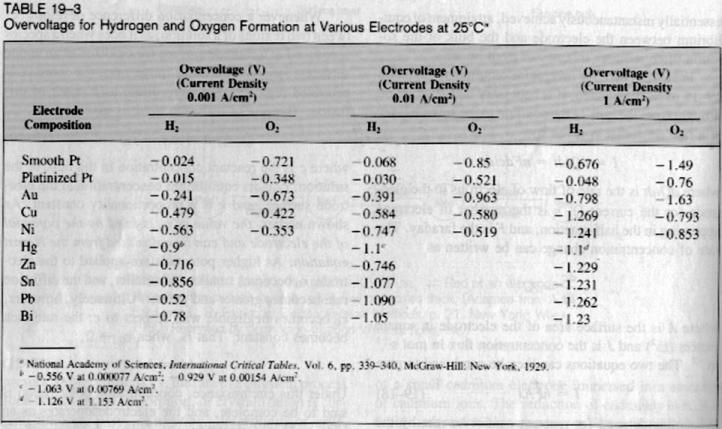
You were already told by Vitus that the Cu electrode can’t not work, just a couple of posts above, so why wasting your material and time?
| Quote: | | 3. The voltage was too high, as SWIM asked in his posts before what is the perfect voltage (aprox.)? |
You seam to talk about the potential about the two working electrodes, don't you? Obviously that is irrelevant. That potential only depends on the
resistance of the cell: I = U / R. You know, exactly like you were told in school. So if you want a higher current you need to provide a higher
potential. This potential has nothing to do with the chemical reaction provided that it is more then the reduction potential (somewhat more than 1V).
The only side effect is the heating of the electrolyte (read the posts above). The potential of the cathode can only be measured in relation to a
reference electrode and since you seam not to use one it is useless to talk about any voltage at all. Rather refer to current per surface which is the
most important electrical factor (just check the table for the large influence of A/cm2 on the overvoltage of Pb electrodes!).
| Quote: | | SWIM knows it depends on the H2 generation, but on the other side there is a difference if I use 1.3V or 13V... |
The less H2 evolves the more efficient the reduction is, but since there are other factors besides efficiency that will affect the yields one does the
reduction at somewhat higher currents to speeds things up and reduce some side reaction.
| Quote: | | One more thing. It needs to be verified the P2NP is P2NP, else all experimentation is useless. |
Ever heard about recrystallization followed by melting point measurement?
I think it would help you if you would read the basics on electrochemistry. Also, why the hell do you use plain “P2NP”? Surely you know its
reduction results in a controlled substance. That is a sure ticket to get into troubles. Can’t you get any other beta-nitrostirene?
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
d0c
Harmless
Posts: 9
Registered: 4-8-2006
Member Is Offline
Mood: No Mood
|
|
Nicodem: Thank you for your post.
SWIM did crystal. and mp measurement. Just wanted a secondary validation method.
SWIM posses a license for 3 months for a project that allows production of the reduced P2NP in small amounts, as far as the product is given away at a
specific collection point for proper and authorized elemination!
Anyway, all experiments are conducted in mmol sizes and until now it hasn't even worked.
SWIM was searching for that table in all his chemistry books! Thanks a lot.
Why SWIM asked about the potential was, there ARE reactions which inhibit other reactions at different voltages and in the book "amphetamin synthesis"
they are talking about 12V, other literature gives data of 6V and it was irritating.
[Edited on 6-8-2006 by d0c]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The truth is that voltage is quite irrelevant so long as it exceeds the voltage required for hydrogen evolution ,
it is the current passed through the cell which does the work . I have used triac phase modulated AC fed to a full wave bridge rectifier and then to
the cell where 120 Hz pulses of DC at levels of 100V peak are used for the
electrolysis , and entire cell hums like a transformer from
the chopped waveform .....and electrolytic reduction proceeds just as well , perhaps even better than using a constant DC source . It is the Average
Current that is
the important thing .
I used to laugh sometimes about my electrolysis cell making all the powerlines and transformers in the whole neighborhood start buzzing from the
non-standard loading
I would put on the power grid using my cell which I lovingly referrred to as Dr. Frankensteins Atomic Hydrogen Reactor
[Edited on 6-8-2006 by Rosco Bodine]
|
|
|
d0c
Harmless
Posts: 9
Registered: 4-8-2006
Member Is Offline
Mood: No Mood
|
|
Based on the table above an experiment has been done using a Ni-Cathode and a graphit Anode in H2SO4, GAA,Ethanol Solution with a very small quantity.
Due to the nearly used up graphit electrode the experiment could not be brought to an end. About 3-5A were beeing used.
After isolating the free base using Chloroform and addition of dil. H2SO4 in IPA a crystaline precipitation occured.
Since the amount of material used was analytical < 50mg a calculation was very difficult, leading to overacidation of the IPA solution, making the
crystalline mass disappear rapidly again.
Is it due to the formation of the hydrosulfate salt or how is the overtitration and the redissolving of the crystals explainable???
However, experiments have shown, that Cu Ions seem toxic for this reaction, leading to formation of a polymer.
Nickel seems to be a suitable, but probably not perfect cathode material.
|
|
|
Vitus_Verdegast
Hazard to Others
Posts: 297
Registered: 5-12-2004
Location: Ottoman Empire
Member Is Offline
Mood: tea time
|
|
Rosco Bodine:
Apparantly H2SO4 can be used in an EtOH/AcOH mixture for the reduction of unsubstituted P2NP, if we may believe this reference:
from https://synthetikal.com/Rhodiums_pdfs/chemistry/amphetamine....:
1 mol of phenyl-2-nitropropene, C6H5CH=C(CH3)NO2, is dissolved with a solvent prepared by mixing 1000ml of ethanol with 500ml of acetic acid and 500ml
of 12 N sulfuric acid. The resultant solution is placed in the cathode compartment of a divided electrolytic cell containing a metallic cathode of
mercury, copper, or other metal of similar nature. Current is passed, using a current density of ~0.2 amp/cm2 of cathode surface. The temperature is
kept at about 40°C during the electrolysis which is continued until at least eight Faradays of electricity have been passed.
When the reduction is completed, the amphetamine may be separated from the solution. A convenient way of doing this is by removing the ethanol and
ethyl acetate present by evaporation and then making the residual solution strongly alkaline by addition of caustic alkali. The basic layer thus
formed is separated from the aqueous solution and contains the desired amphetamine freebase.
135 g (1 mol) of amphetamine freebase were stirred into 300ml of acetone in a 1000ml erlenmeyer flask. To the resultant solution there were slowly
added under constant agitation 115.3 g of 85% phosphoric acid (containing 1 mol of H3PO4), care being taken to avoid any sudden rise in temperature or
local overheating due to the considerable amount of heat that is evolved. During the addition of the phosphoric acid a fine, white, flocculent
precipitate appears which becomes more and more dense and abundant, as the quantity of added acid increases.
When the entire quantity of the phosphoric acid has thus been added, agitation of the mixture is continued for about a half-hour or more to insure
complete conversion. The precipitate is then allowed to settle, the supernatant liquid is drawn off, and the residue is filtered. The precipitate thus
separated is washed with acetone and is then dried by evaporation to constant weight. It forms a fine, white, impalpable powder consisting of pure
monobasic amphetamine phosphate.
Reference: Pharmaceutical Manufaturing encyclopedia (1988)
Sic transit gloria mundi
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Interesting . That appears to be a much more recent reference . The experiments which I was doing was thirty years ago and strictly on a lead
cathode .
If I had pursued my experiments further , a mercury pool cathode was exactly where I was going next . The whole reaction would be cleaner I think
than using a lead cathode , which always corroded , causing some sludge in the cell . It may be that H2SO4 was avoided in the case of the lead cathode
for some reason , I honestly can't remember , but I know I used HCl instead of H2SO4 for some reason .
Never tried phosphoric acid , never even heard of this being a common form , but you would need a food grade
phosphoric which would be a lot more expensive than electrolyte grade sulfuric which is exceptionally pure and cheap .
In a two liter volume , it would still be a slurry I would guess as the product is accumulates towards the end of the reduction . It probably does
precipitate in the electrolyte , but then as the alcohol is evaporated and the relative concentration of water is increased , the precipitate
redissolves in the residual water , ready for the freebasing . The yield is probably good if this was referenced from a commercial process .
|
|
|
Vitus_Verdegast
Hazard to Others
Posts: 297
Registered: 5-12-2004
Location: Ottoman Empire
Member Is Offline
Mood: tea time
|
|
Corrosion also occurs always in my experiments, H2SO4 or HCl, I can use the cathode only once for a reduction.
Attempts to electrolytically coat such a used corroded lead electrode always results in a thick white crust of lead sulfate being formed instead of
the usual layer of PbO2.
I have had very good results and apparantly less corrosion using a lead amalgam cathode. The mercury was applied electrolytically, but I remember
reading somewhere that the preparation of a lead amalgam electrode is as simple as rubbing the clean lead surface with metallic mercury.
Amphetamine phosphate is available through pharmaceutical supply houses where I live. It has been claimed IIRC to be absorbed somewhat slower into the
bloodstream than the sulfate, although this difference is probably marginal.
There is not much water present here, maybe the benzedrine sulfate dissolves well in the mixture of AcOH/12N H2SO4 ? Many amine salts are known to
redissolve when an excess of acid is present, when eg. precipitating from an alcoholic solution.
Sic transit gloria mundi
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Probably could do the amalgamation with about 6% nitric acid containing mercuric nitrate . Would be easy enough to dissolve the mercury in a bit
excess of d 1.4 nitric and dilute to something like 6-10% strength . Should work likewise on a few other metals copper , nickel ,...... perhaps
titanium .
|
|
|
Ullmann
Hazard to Self
Posts: 51
Registered: 22-12-2004
Member Is Offline
Mood: No Mood
|
|
Is there any practical advantage of using a mercury cathode instead of a lead cathode?
As judged looking at the table posted above the overvoltage of lead is equal or higher than mercury for current density above 0.01 A/Cm². Practically
what does that mean in our case? Will a mercury cathode perform better than a lead cathode?
Rosco, you said mercury would not corrode as much as lead, but will it still corrode a bit giving Hg salt contamination in the catholyte? I seem to
remember that to make mercury sulfate one need to heat mercury in concentrated sulfuric acid to more than hundred degrees temperature. To make mercury
acetate, one has first to put some nitric acid to catalyse the oxydation of mercury before the nitrate gets displaced by acetate ion. Because here we
have low to moderate temperature as well as reducing medium i do not think mercury will dissolve in this case. But i can only assume lead is more
easily oxydized than mercury in acid medium if i read your comments. Also vitus report improvement using amalgated lead... Will some of the mercury
cathode dissolve or will it be totally inert?
Also how can anyone suggest a good setup using this liquid electrode? How much Hg would be needed for a 50 mmol batch for instance? Is the Hg standing
at the bottom of the catholytic compartiment or is a dropping mercury drop used?
Thanks
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Honestly I don't know if the lead or mercury is better ,
I only worked with lead and it was 30 years ago and
I don't have the notes or articles anymore from what
I was doing then . All I remember , IIRC is that such
reductions had been done by some workers using
a mercury pool cathode , and other than lead that
is all I remember in the way of cathode materials which
had been reported useful . I am pretty sure that
it was my thinking at the time when observing the
level of erosion on lead cathodes , that this is simply
one of the more nasty electrolytic reductions where
a side reaction is probably occurring that involves attack
of the electrode , by the regional oxidation of the metal
itself where it is not being provided any cathodic protection by the evolution of nascent hydrogen , but
is being attacked oxidatively by the nitro group and
electrolyte actually oxidizing the lead , similarly as if an acid plus a metal was being used as a chemical generation of nascent hydrogen , with the
metal going into solution as the acid salt and being consumed in the process .
Probably there is an optimum pH and temperature
and current density where this corrosion of the cathode
is minimized .....but that would require some extensive
process variations to determine what is optimum .
Basically all I did is observe the cathode and crank up the current until I could see a little effervescence of hydrogen , back off to minimize it
without it disappearing
completely .....and call that about right for current density , adjusting it periodically whenever the cell
would go to bubbling freely again as the reduction proceeded . So the current density has to be ramped
downward as the reduction proceeds further towards
completion .
|
|
|
stoichiometric_steve
National Hazard
Posts: 819
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by Vitus_Verdegast
Use a heat gun (for paint removal) or similar to soften the outer plastic case before attempting to cut it. |
how is it possible to open a car battery without risking an electrical shock?
|
|
|
Mr. Wizard
International Hazard
Posts: 1042
Registered: 30-3-2003
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by stoichiometric_steve
| Quote: | Originally posted by Vitus_Verdegast
Use a heat gun (for paint removal) or similar to soften the outer plastic case before attempting to cut it. |
how is it possible to open a car battery without risking an electrical shock? |
It's quite tough for a 12 volt battery to push enough amperage through normal skin to give you a shock. That said, it can send some current through
wet or sweaty skin or from any device that has a significant inductive 'kickback' voltage, such as a car horn in operation. The actual danger is more
from the chemicals, cutting yourself while wrestling with the tough plastic. or getting a nice high current short from accidentally cutting into two
plates of a cell still charged up. This will make sparks and heat, and may splatter acid
I'm sure everyone knows that a charged battery has more sulfuric acid in the electrolyte than an uncharged one, so if you want more acid charge it up.
This will convert the lead sulfate in the plates to 'Lead Peroxide', a dark brown substance, on the positive plates, and 'spongy lead' on the negative
plates.
Has anyone done any battery rebuilding with the new plastic cased batteries? I realize it was done all the time with the old hard rubber cases and
poured pitch tops. It looked like a real dirty job. I'd be interested in hearing about a real good way to open up a plastic battery in a way that
would allow rebuilding it. I have a nice scanned book somewhere on rebuilding the old ones.
|
|
|
stoichiometric_steve
National Hazard
Posts: 819
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by Mr. Wizard
I'm sure everyone knows that a charged battery has more sulfuric acid in the electrolyte than an uncharged one, so if you want more acid charge it up.
|
didnt really think about that...good point 
so i guess i'll just let the battery run flat, because i have no real interest in the electrodes rather than the diaphragm bags separating the
electrode compartments.
anyway thats a shame since i was about to buy a new one.
so the way to do it for a fully charged, new battery is to just avoid touching the connector terminals and the electrodes? i'm gonna wear rubber
gloves anyways, so the electric shock doesnt really pose a great risk.
another interesting for anybody attempting the reduction with the cell according to Vitus: from the GB patent mentioned above i gathered that the
addition of hydroxylamine salts to the catholyte greatly improves yield.
it's been a while since i have done electrochemically monitored titration at the univ. but i remember something about oxime titrations, where the
excess hydroxylamine prevented or induced some reaction at the electrodes, what would that be? i guess it can't be reduced to ammonia at the
cathode...and that means something...
furthermore,
when using an ATX PC power supply for the electroreduction, does it make more sense to use the 5V or the 12V line? in both cases, the potential
between the electrodes should well exceed the potential needed for reduction of the substrate, while the 5V line can provide much more Amperes than
the 12V line.
is it important to use equally sized anodes and cathodes, or can electrodes of differing sizes be used?
[Edited on 14-7-2007 by stoichiometric_steve]
|
|
|
12AX7
Post Harlot
Posts: 4803
Registered: 8-3-2005
Location: oscillating
Member Is Offline
Mood: informative
|
|
How much voltage do you need? (I haven't been watching this thread.) I'm going to bet it's not more than 5V. In the 1-4V range, you'll need a
ballast resistor from the 5V supply to keep it to a comfortable rate and within ratings.
Tim
|
|
|
stoichiometric_steve
National Hazard
Posts: 819
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by 12AX7
How much voltage do you need? |
the reduction proceeds beyond -1.1V, so i guess just letting it run at 5V will do fine. i have no idea how to manipulate the voltage...
some people use 12V, some (like Rosco) use pulsed 100Hz DC, which seems to work fine, too.
my only concern is the higher amps that the 5V line can provide, which would affect current density and shorten the time for completion.
|
|
|
12AX7
Post Harlot
Posts: 4803
Registered: 8-3-2005
Location: oscillating
Member Is Offline
Mood: informative
|
|
Power supply outputs are current ratings. Your house's water hookup might be a hundred gallons per minute but that current obviously isn't flowing
all the time. Same thing here.
Note that 5V is a lot for an electrochemical cell, so a large current may flow anyway. Which is why I suggested a ballast resistor. Look it up!
Tim
|
|
|
stoichiometric_steve
National Hazard
Posts: 819
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
Patent GB2122617 makes use of hydroxylamine salts in conjunction with a zinc plated lead electrode to reduce nitroalkenes to saturated amines in
[claimed] good yield.
This document from the Rhodium archive says that the presence of hydroxylamine due to hydrolysis of oximes represses the reduction to the amine by
decreasing hydrogen overvoltage at the cathode.
it seems to me that this is quite some contradiction.
why would one add hydroxylamine to the catholyte while it is stated in the other document that this inhibits the complete reduction of the substrate?
something else (I):
i just cracked a car battery open this afternoon, let me tell you that its absolutely vital to take the TOP off, and dont EVER use a worn out or used
battery, because you will spend the money on cleaning the place where you took the battery apart. theres such an incredible amount of sludge (PbSO4,
PbO2?) to deal with which of course needs special care/disposal. dont flush this shit down the drain...
something else (II):
the design for the electroreduction cell as proposed in the first post of this thread is nice, but not really ideal, since the diaphragm bag doesnt
allow for magnetic stirring.
the diaphragms from car batteries can be cut to pretty big pieces, about 12cm by 20cm. fitting this in a frame dividing a larger cell would be much
better, so you end up with 2 stirrable compartments which can be cooled by spiraled plastic tubing submersed in the electrolyte.
does anybody have a reference design employing this type of diaphragm?
[Edited on 8-8-2007 by stoichiometric_steve]
[Edited on 8-8-2007 by stoichiometric_steve]

|
|
|
Vitus_Verdegast
Hazard to Others
Posts: 297
Registered: 5-12-2004
Location: Ottoman Empire
Member Is Offline
Mood: tea time
|
|
| Quote: | Originally posted by stoichiometric_steve
Patent GB2122617 makes use of hydroxylamine salts in conjunction with a zinc plated lead electrode to reduce nitroalkenes to saturated amines in
[claimed] good yield.
This document from the Rhodium archive says that the presence of hydroxylamine due to hydrolysis of oximes represses the reduction to the amine by
decreasing hydrogen overvoltage at the cathode.
it seems to me that this is quite some contradiction.
why would one add hydroxylamine to the catholyte while it is stated in the other document that this inhibits the complete reduction of the substrate?
|
Thanks for providing that patent. The contradiction beats me too. The patent recommends solutions greater than 0.1M hydroxylamine as the catholyte.
"The chemical efficiency increases with increasing concentration of the hydroxylamine. The upper limit of the hydroxylamine concentration is
determined by the solubility in the catholyte."
Towards the end of a nitroalkene electroreduction I always noticed increasing H2 evolution. Always chalked that one up to decreasing overvoltage too.
They don't seem to say exactly why hydroxylamine salts should be beneficial.
Still it's certainly worth a try. Interesting also is the fact that they claim having reduced 1-(3-indolyl)-2-nitropropene in 60% yield.
This patent uses a filter-press electrolytic cell. The attached patent provides a good description of such a cell. It lacks a suitable cooling system,
which should be inserted between every cell compartment
[Edited on 9-8-2007 by Vitus_Verdegast]
Attachment: US4490231.pdf (763kB)
This file has been downloaded 1307 times
Sic transit gloria mundi
|
|
|
Vitus_Verdegast
Hazard to Others
Posts: 297
Registered: 5-12-2004
Location: Ottoman Empire
Member Is Offline
Mood: tea time
|
|
| Quote: |
the design for the electroreduction cell as proposed in the first post of this thread is nice, but not really ideal, since the diaphragm bag doesnt
allow for magnetic stirring. |
Why stir the anolyte in the diaphragm bag? But I know the design is rather crude and should be improved.
[Edited on 9-8-2007 by Vitus_Verdegast]
Sic transit gloria mundi
|
|
|
stoichiometric_steve
National Hazard
Posts: 819
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by Vitus_Verdegast
Why stir the anolyte in the diaphragm bag? But I know the design is rather crude and should be improved.
[Edited on 9-8-2007 by Vitus_Verdegast] |
actually, the design is pretty ghetto and thus lovely. i was under the impression that the anolyte could well use some cirulation, too. maybe just to
ensure good cooling...so if its not necessary, all the better!
|
|
|
| Pages:
1
2
3
4 |











