| Pages:
1
2
3
4
5 |
smuv
National Hazard

Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Benzamide...
Unhappy with the yields from the previous benzamide synth, I investigated different catalysts for the amidation of benzamide in a urea melt. In
practice I did a shitty job controlling some variables; therefore, I don’t really know how reliably my results can be compared to one another.
Benzoic acid synthesis
224g Potassium Benzoate
422mL water
142mL 31.45% HCl solution
The Potassium Benzoate was dissolved in warm water and HCl was added. Benzoic acid immediately precipitated out, and the solution was allowed to cool
to room temp. The crude benzoic acid was collected via vacuum filtration, using the filtrate to complete the transfer and the filter cake was sucked
fairly dry. The crude benzoic acid was dissolved in 3.5L of boiling water (a very little bit steam distilled creating very beautiful crystalline
benzoic acid snow!), the vessel was insulated and allowed to slowly cool overnight. The solution was vacuum filtered, using the filtrate to complete
the transfer, washed with ca. 250-300mL water and sucked dry. The benzoic acid was dried at 150-200F in an oven for about an hour.
The entire procedure was done 2x in parallel yielding ~300g of benzoic acid.

(300g of recrystallized benzoic acid)
Benzamide synthesis
I tried three different catalysts for the synthesis of benzamide from urea and benzoic acid. I used a phosphoric acid catalyst, a boric acid
catalyst, a zinc oxide catalyst and an uncatalyzed control. The uncatalyzed, phosphoric acid catalyzed and boric acid catalyzed processes for
formation of amides are known to literature, however I wanted to give the zinc oxide catalyst a try because I thought it might work well for this
reaction.
Boric acid catalyzed reaction
90g Benzoic acid
133g Urea
6.9g Boric acid
Phosphoric acid catalyzed reaction
90g Benzoic acid
133g Urea
1.75mL Phosphoric acid
Zinc oxide catalyzed reaction
90g Benzoic acid
133g Urea
11.8g ZnO
Uncatalyzed reaction
30g Benzoic acid
43.7g urea
All reactions were performed in parallel; the 3 catalyzed reactions were run in 500mL Erlenmeyer flasks while the uncatalyzed reaction was run in a
250mL filter flask. The flasks were covered with aluminum foil and heated for 2.5 hours on a sand bath. The sublimate was tapped down from time to
time. I tried to maintain the temperature of the melts at 180c, but because I left the reactions more unattended than I should have, I did a poor job
at this. Additionally, the temperatures varied a good deal melt to melt, which made things tricky. About 33% through the reaction, the phosphoric
acid and zinc oxide catalyzed melts were running at 210c, the uncat. was 205c and the boric acid was 185c. Later I undershot the temperatures, and
they hovered around 150c. For the last hour I got the temps a little closer around 190-200c.

(The sand bath...)
After 2.5 hours, the melts were poured into empty Griffin beakers and allowed to cool to just above 100c. To the 3 catalyzed reactions 500mL of water
was added and to the uncatalyzed, 150mL of water was added. The solutions were boiled with stirring and a 28% solution of ammonia was added (35mL for
the cat, 10.5mL for uncat). Next the solutions were cooled to just below room temp and vac filtered. The filter cakes were crushed up and dried in
an oven at about 150F.
To purify the benzamide, the idea was to extract the crude products with boiling xylol, filter out the undissolved junk and allow the benzamide to
crystallize out upon cooling. I tried this for the phosphoric acid cat. reaction two times. Both times the benzamide immediately crystallized
clogging the filter funnel. I believe to efficiently do this a heated filter funnel would be required and/or a very large amount of xylol would need
to be used. I was able to recover most of the product (I think) by allowing the very little xylol that passed through the funnel to cool, and
collecting the precipitate by vac filtration. The whole filtration apparatus was washed with warm denat. ethanol to recover most of the benzamide
which had clogged it. I took what had precipitated on the filter paper and dissolved this in hot ethanol as well; both alcohol solutions were
combined, boiled, allowed to cool to ca. 35c and vac filtered. The ethanol was then slowly evaporated in a gently heated casserole dish. The
precipitated benzamide was collected and dried in an oven at 150F. The other reaction products were extracted with ethanol (250mL for the ZnO, 325mL
for Boric acid, 150 for uncat), filtered, cooled to ca. 35c and the solvent was evaporated.

(Melt from boric acid catalyzed reaction dissolved in boiling water)

(Water recrystallized, boric acid catalyzed, reaction product)

(After filtering and cooling the warm ethanol extracts benzamide slowly precipitated)

(Product recovered from phosphoric acid cat reaction: precipitate after evaporating ethanol on left, extract from boiling xylol on right.)
Yields
Phosphoric acid cat. 36.8g (precip. from xylol + precip. from EtOH) --> 41% yield
Boric Acid cat. 73.3g --> 82% yield
ZnO cat. 32.8g --> 37% yield
Uncatalyzed 18.1g --> 61% yield

(final pure(ish) products)
I didn’t perform any melting point assays, but visually the products from the ZnO and uncatalyzed reaction looked a fair bit darker than the
phosphoric and boric acid catalyzed reactions. The portion of phosphoric acid catalyzed reaction product that precipitated from xylol was not
crystalline but was a very clean white powder.
Discussion
Benzoic acid synthesis
I made a mistake with this synthesis, I found out only after the fact that the HCl I was using was 31.45% instead of 34% as I had assumed; therefore
instead of using a 10% excess of HCl I only used the stoichiometric amount. I hope this is the reason for the lowish yields for this simple
preparation. Of course a percentage was also lost in the recrystallization.
Benzamide synth
The results are all over the place. Throughout the reaction the boric acid catalyzed reaction stayed closest to the 180c target which could explain
the higher yields. It is possible that a lot of benzamide was lost in the phosphoric acid catalyzed reaction with the xylol mishaps. I think it is
fair to bet that ZnO doesn’t do a good job catalyzing this reaction at the temps used, it is possible that at lower temps it may be more effective.
The only concrete conclusion that I can come to, is that boric acid does a pretty good job as a catalyst.
I think I got higher yields with the boric acid catalyzed reaction than in my previous experiment, because I recrystallized the product from boiling
water. While this did not really remove all the impurities because I never filtered the solution hot, I think it allowed the benzamide to precipitate
out of the solution as distinct crystals rather than stay trapped in the solidified melt. I believe the ethanol extracted product could be made very
pure by recrystallization from xylol. Also simply recrystallizing from hot water (in my hands) did not provide a sufficiently pure product, as even
the water recrystallized product from the boric acid catalyzed reaction, which dissolved completely in boiling water did not fully dissolve in boiling
ethanol.
Conclusion
As a comparison of different catalysts for the synthesis of amides from carboxylic acids, I view this experiment as a failure. On the other hand, for
the production of a fair bit of benzamide (albiet less than I had wanted) this series of experiments was successful.
Notes:
I have been using the term catalyst loosely, for the phosphoric acid and boric acid catalyzed reactions, precatalyst would probably be a better term.
The idea for the ZnO catalyst came from research into the synthesis of ethylene carbonate from urea and ethylene glycol; in this reaction ZnO
catalyzes the formation of ammonium cyanate. Additionally, I thought ZnO’s weak acid and weak base sites might help stabilize the various
transition states required to form the amide.
That’s all…
[Edited on 12-1-2008 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Regardless of the lossy variable control in your parallel experiments, I think your work does give quite some useful data on this reaction. There is
not much about amide formation with urea on this forum and yours are great contributions to this simple method of COOH->CONH2 transformation.
Thanks!
If you have time enough it would be nice if you would do a review of your experiments and literature data that you gathered during your work, and post
it in the Prepublication section.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Nice work Smuv, thank you for taking time to share this with us!
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Thank you both for your appreciation.
@ Nicodem I may one day post something in prebulication about this process, but for now my research of this reaction is not complete. I would be
interested to hear of your experience with the boric acid catalyzed reaction (substrates and yields if you still have the data).
Mechanism
The role ammonium cyanate plays in this reaction was not immediately clear to me, here is the mechanism; it is interesting IMO.
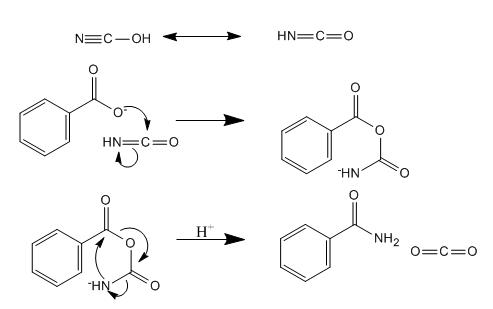
Source: Molecules 2006, 11, 318-324
[Edited on 12-2-2008 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
Taoiseach
Hazard to Others
Posts: 241
Registered: 16-3-2008
Member Is Offline
Mood: No Mood
|
|
Awesome work smuv, thanks for sharing!

|
|
|
Alchemist
Hazard to Self
Posts: 93
Registered: 22-6-2002
Location: Hostton Texas
Member Is Offline
Mood: No Mood
|
|
Urea Acidolysis
Hello all,
Beside the 3 examples that where given in "Examples of Urea acidolysis" , Formic Acid, Acetic Acid, Benzoic Acid with Urea to make Formamide,
Acetamide, and Benzamide, does anyone know of others that work with Urea? If so do you have information you can add to the list?
For example would Oxalic Acid and Urea make Oxamide?
Thanks the Alchemist.....
[Edited on 23-3-2009 by Alchemist]
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
The Hoffman Elimination reaction is the most difficult part of this type of synthesis. Nice work with this synthesis. I have used this reaction to
make anthranilic acid from phthalimide. The only problem is that anthranilic acid is difficult to precipitate in high yields du to the fact that it is
an amino acid which has a narrow isoelectric point over which it will precipitate. This method should be applicable to phthalic acid diamide for the
synthesis of o-phenylenediamine. The use of TCCA is extremely interesting, it keeps the reaction volume low for easy work up. This may be applicable
to making 3-aminopyridine from nicotinamide.
Amateur NMR spectroscopist
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
@alchemist: if by urea acidolysis you mean the formation of amides by cooking them up in a urea melt, then yes there are many substrates which this
application is applicable to. The only thing that would inhibit a carboxylic acid from reacting that I can think of is if the acid had other
functional groups that also reacted in the urea melt. I had wanted to try comparative examples between dichloro, trichloro and acetic acid a few
months ago, but my synthesis of trichloro and dichloroacetic acid failed...
@benzylchloride1 the reaction volume can likely be kept even lower if the the formation of the formation of the N-chloramide were done in methanol.
In methanol this chlorination goes like a shot because the cyanuric acid is insoluble in MeOH. The hoffmann rearrangement in methanol unfortunately
gives rise to urethanes. I bet though, addition of the solution of the N-chloramide in MeOH to a NaOH solution would yield the amine. An attempt was
made w/ urea --> hydrazine a while ago, but it didn't work out (not really a model substrate). An attempt was planned to try this on a
dihydrocinnamide but the formation of the dihydrocinnamic acid did not go as planned. More work on this rxn is planned eventually, although for now,
time is scarce.
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
I am currently running the benzamide synthesis. I heated 90g of benzoic acid, 133g urea and 6.9g boric acid in a 500 mL flask. The mixture was heated
at 165 c for 2.5 hours. The product was poured into a glass dish and broken up. The product was well mixed with water and filtered. The filtrate was
acidified to recover unreacted benzoic acid. Around 25g precipitated out estimated. The crude benzamide is currently covered with methanol and will be
heated to boiling, filtered from urea condensation by products and then concentrated with a rotary evaporator. I should have a melting point, and a
percentage yeild within 2 days.
Amateur NMR spectroscopist
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Did you dissolve the melt and recrystallize from a dilute boiling ammonia solution? This IS necessary to get the best yields as much product stays
trapped inside the melt; methanol cannot dissolve benzamide that is surrounded by biuret.
That being said, I am glad to see someone repeating one of my experiments!
[Edited on 3-24-2009 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
Ebao-lu
Unregistered
Posts: N/A
Registered: N/A
Member Is Offline
|
|
Nice, smuv! Boric acid seems to be a versitile catalyst for -CO(X) activation. And how did you get rid of biuret? What is the purpose of using dilute
NH3 solution(to remove biuret)? Biuret is also poor soluble in cold water, and moderately soluble in ethanol, so it is possible some biuret is still
there(not sure). Try to dissolve the rest products (ZnO, B(OH)3, uncatalysed) in boiling xylene to see if there is some crap
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
I got a very low percentage yield around 27% after recrystallization from methanol. Only a small amount of urea condensation product was obtained
after filtering. Upon heating the product with aqueous NaOH, a strong odor of ammonia was apparent, due to the hydrolysis of the amide.I ground the
crude fusion product in a morter and pestle before recrystalization. The product melted at 120 degrees Celsius. The product must be contaminated with
benzoic acid due to the low melting point. Upon acidifying the aqueous filtrate, a large quantity of unreacted benzoic acid was recovered. The mixture
did not get hot enough during the fusion, 165 degrees Celsius. I am currently producing aniline from the benzamide I obtained. I steam distilled the
aniline out of the reaction mixture, salted out and extracted the aniline with toluene. The toluene extract is now drying over NaOH. Alot of oily tar
was produced in the reaction, lowering the yeild. How could the tar formation be minimized?
Amateur NMR spectroscopist
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
I think you should read my experiments and feedback I received from other members as it seems like you are repeating some of my mistakes.
Methanol is a really bad solvent for recrystallization or purification of benzamide as benzamide is very soluble in it as well as some of the
condensation products formed from urea. This could explain your poor yields.
If you heat the crude melt in boiling dilute ammonia water, all benzoic acid as well as many other impurities are removed.
The low mp is not due to benzoic acid, it is due to benzonitrile.
This being said, In my hands I got variable results w/ this amidation.
For the purposes of the hofmann rearangement, most impurities but the condensation products of urea will not interfere w/ the reaction; for this
reason rigorous purification of the benzamide is IMO unnecessary. For the Hofmann rearrangement, very good cooling, stirring and slow addition of the
amide are absolutely paramount! I had previously stated that the addition of the amide does not appear to be exothermic; on large scales it is, and
allowing the temp to rise even to 15c markedly reduces yields. This being said, I did get some tar formation every time during this synthesis.
There is 1 modification you may wish to try if you repeat (I had planned to try it). Make a sol of .33-.45 eq of TCCA and add the amide to this
slowly w/ stirring and rigorous cooling. If the amide does not go into sol, add 1 eq of NaOH. Once the amide goes into sol, then add the rest of the
NaOH (6 eq in total IIRC) slowly with very good cooling. Allow the sol to slowly come to RT after addition, then heat slowly to ~75c.
By this method you are first generating the N-chloroamide w/ less risk of degradation of the chloramide to benzoic acid and ammonia (which I believe
is a competing reaction).
p.s. I am glad to see someone try this reaction, and hope that w/ further experimentation you work out some of the problems you have encountered. I
also know all to well, it is very easy for someone to find flaws in something you have done after the fact. I hope you are not discouraged and the
second time around can improve the yields.
p.p.s. In my hands tar formation did not seem to significantly impact yields. Although I vaguely recall more tar formation in a large scale reaction
where I added my amide too quickly and consequently allowed the temp of the solution to rise.
[Edited on 3-30-2009 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Amides from Esters
Preface
To further research amidations in molten urea, the synthesis of benzamide from methyl benzoate performed.
Methyl Benzoate
This is straight from vogel...
30g Benzoic acid
100 mL Methanol
~3 mL Sulfuric acid
Refluxed 4.5 hours. Allowed to stand overnight. Separated ester, extracted aqueous phase w/ CH2Cl2. Combined Organics, washed with bicarbonate and
water. Dried over CaCl2. Removed solvent via distillation.
Obtained 10.9g of a clear just slightly yellow oil.
Benzamide
10.9g Methyl benzoate
11g Urea
.75g Boric Acid
Mixed everything, heated in oil bath for 2 hours. Oil bath temp was increased from 140c to 170c over course of rxn. At first yellow plates
precipitated but with further heating heating a lot of brownish colored trash precipitated. By end 2h only ca. 4mL of molten stuff remained, the rest
was solid. The melt was allowed to cool and 125mL of 10% NH3 solution was added. Solution was boiled with stirring for ~10min and allowed to cool in
a water bath. About 3mL of oily methyl benzoate remained unreacted. The solution was vac filtered and obtained off-white powder was dried in oven.
Yield 2.95g (30%) of a fairly clean product.
Discussion
The yields of the esterification are probably low because the sulfuric acid used was drain cleaner, and likely contained a fair bit of water.
For the amidation, in the future more urea should be used, because a lot of methyl benzoate was left unreacted, and would have had no chance to react
if the rxn was extended longer because most of the urea was decomposed after 2h. Also less ammonia solution should be used in work-up as the
solubility of benzamide in water is 1g /~75mL. Additionally unreacted methyl benzoate could have dissolved some of the benzamide as well.
Despite these shortcomings, the purpose was to determine if esters react well in this reaction, it appears with some subtle modifications, this
process can be applied to esters if so desired. Of course there are other ways of making amides from esters, some are more convenient. However, this
is just one more choice for the amateur chemist.
[Edited on 5-10-2009 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
Allowing the ester to stand overnight in the presence of the acid could have contributed to hydrolysis of the ester, lowering the yields. I have
conducted the methyl benzoate synthesis several times and always got above a 70% yield after distilling the ester. I used 96% ACS grade H2SO4, I only
use this acid for esterifications and other synthesis where water content is an issue. The 92% drain cleaner can be concentrated by distillation if
suitable apparatus and a hood is available. The ammonolysis with the formation of the benzamide is best conducted at room temperature. At high
temperatures, basic hydrolysis is a competing reaction. Ammonium benzoate will be the predominate product. I have prepared acetamide from ethyl
acetate in this manner; the amide is isolated by distillation.
Amateur NMR spectroscopist
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
| Quote: | | Allowing the ester to stand overnight in the presence of the acid could have contributed to hydrolysis of the ester, lowering the yields.
|
Edit: Actually you are right, for the synthesis of ethyl acetate, the equilibrium constant is 3 at room temp and 4 at 100c so this could have reduced
yields.
| Quote: | | The ammonolysis with the formation of the benzamide is best conducted at room temperature. |
Eh you missed the whole point of my post.
The product after the urea melt was not heated with ammonia solution to form benzamide, this was done to remove impurities (namely benzoic acid and
biuret) from the crude product. It is really something you should have done in your synthesis, it would have increased the yields of your alcohol
extraction by allowing the benzamide to re-crystallize free from biuret and other condensation products and thus accessible to solvation by the
alcohol.
[Edited on 5-12-2009 by smuv]
[Edited on 5-12-2009 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
I am sorry that I misread your post. Using urea to form an amide form an ester is a new reaction for me. I have always used ammonia for forming amides
from esters. I plan on running this synthesis again using your modifications that produce the highest yields of product. My aniline from my first
synthesis would not totally dissolve in concentrated hydrochloric acid. All of my ammonialysis reactions with aqueous ammonia proceded in high yields.
I really want to synthesize around 100ml of aniline by this method; I have benzene, but the nitration and reduction require chemicals that are some
what costly for me. In a dilute aqueous acidic solution, ester hydrolysis would be the predominate reaction.
Amateur NMR spectroscopist
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
| Quote: | | Using urea to form an amide form an ester is a new reaction for me. |
Now you understand why i tried it. 
Via the urea melt method, you could easily make that quantity of aniline (provided you have a 3L flask for the hoffmann rearrangement). Read my posts
above, I gave tips regarding steps to take for larger scale hoffmann rearrangements, namely, COOLING!
Also, I am starting to get the impression, that the size of the flask is important. I ran the above reaction in a 250mL flask (too lazy to clean a
100mL flask) and I think in larger flasks the urea might decompose more quickly without reacting with the ester/carboxylic acid.
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
I conducted a benzamide synthesis several days ago. 90g of benzoic acid, 133g of urea and 6.9g of boric acid were mixed together in a 500mL flask. The
flask was heated with a mantle and kept at 180C for 2.5 hours. The product was poured into a glass bowl and transferred to 500mL of water in a 1L
beaker. 100mL of 10% ammonia was added and the mixture was heated to boiling. The solution was then filtered from amorphous byproducts and upon
cooling, benzamide crystals filled the flask. The product was filtered after allowing the solution to cool over night. The product was washed with
water and dried in a 70C oven. 52g of product was obtained, with a melting point of 120C. The product was obtained with a percentage yield of 59%.
Another synthesis was carried out today, and is currently cooling and crystalizing. The mixture was kept between 190C and 210C to see the effect on
yields. More urea condensation products were noted and the mixture had a definite benzonitrile odor. The fume hood works extremely well, the odor was
only apparent when the odor of the mixture was carefully tested. Does anyone know if benzamide could be heated to a high temperature under vacuum to
produce benzonitrile and water as the side product? This could be turned into a working procedure for the synthesis of this compound without using
hard to obtain reagents such as SOCl2, AC20, P4O10, etc.
Amateur NMR spectroscopist
|
|
|
Random
International Hazard
Posts: 1018
Registered: 7-5-2010
Location: In ur closet
Member Is Offline
Mood: Energetic
|
|
Quote: Originally posted by Dr. Beaker  | interesting discussion...
I want to make pentafluoro aniline from pentafluoro benzoic acid.
the procedure posted by S.C. Wack looks atractive to me, however I'm afraid the poorly nucleophilic oxygenes of C6F5COOH would hinder any reaction
that include protonation of the COOH...
the only way I found 'till now that uses (relatively) simple reagents is this:
C6F5COOH ---> C6F5COCl (using SOCl2)
C6F5COCl ---> C6F5COONH2 (by addition of hydroxyl amine)
C6F5COONH2 ---> C6F5NH2 (-CO2 by heating with K2CO3)
any thoughts anyone? |
Does that mean that we can make aniline from benzoic acid and hydroxylamine?
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Its not as simple as that, see: lossen rearrangement.
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
Perhaps this has been pointed out before, but consider the following:
Instead of using the urea as a source of ammonia, why not use it as a means to irreversibly consume water sideproduct (forming ammonium carbamate and
ammonium carbonate).
Perhaps we could try heating ammonium benzoate in a urea melt. Keeping the temperature as low as possible while still having it molten minimizes
formation of biuret, but may still be enough to dehydrate the ammonium benzoate.
Or for that matter, what about molten ammonium sulfamate as dehydrating agent? It's non-acidic and the sideproduct is not organic soluble. Presumably
the hydration to diammonium sulfate is rather energetically favorable. An issue with this would be the higher melting point of the sulfate product,
however, and an excess to keep the melt liquid might go to the nitrile (which would itself be great, but not what's wanted here)
[Edited on 1-15-11 by UnintentionalChaos]
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
For a normal transesterification, the use of a dehydrating agent indeed does push reactions to completion. But if you look at the mechanism for the
reaction given further up the page, you will notice that water is not produced. In this reaction urea does not simply provide a source of ammonia,
instead it serves as a source of isocyante.
|
|
|
Random
International Hazard
Posts: 1018
Registered: 7-5-2010
Location: In ur closet
Member Is Offline
Mood: Energetic
|
|
I figured lossen rearrangement includes messing with isocyanates. Aren't those extremly toxic?
|
|
|
Random
International Hazard
Posts: 1018
Registered: 7-5-2010
Location: In ur closet
Member Is Offline
Mood: Energetic
|
|
I tried today synthesis of benzamide in a test tube. I mixed urea and benzoic acid which filled half of the test tube then I started heating it using
a candle. After some time urea melted and I continued to heat it. I started to notice weak fruity/cat shitty smell, but after some time smelling it my
heart rate became faster and I did get some strange feeling of not feeling right. This was one outside, I stopped heating it and left it outside now.
By the way, I was leading the vapors into vinegar hopefully to neutralize ammonia, but a big amount of vapors escaped from that too, maybe if some
isocyanate was made the vapors were also burned with candle forming HCN?
Edit:
I left it outside and it stopped smelling bad, now I have some crystalline mass with reduced volume, I am not even sure if it's benzamide or ammonium
benzoate or maybe even urea? The mass hardened from liquid phase as I see because it had floating some more benzoic acid on it. Benzoic acid volume
greatly decreased and I think almost nothing sublimated (there is just some little sublimated dust on the test tube walls).
Now, I want to see if I had success with that so I will attempt hoffman degradation using bleach. Do I need to use sodium hydroxide currently? I don't
have it, but maybe Na2CO3 could work too?
I also have some calcium hydroxide too, but the synthesis of NaOH from Na2CO3 and NaOH wasn't success this time.
[Edited on 26-3-2011 by Random]
[Edited on 26-3-2011 by Random]
[Edited on 26-3-2011 by Random]
[Edited on 26-3-2011 by Random]
|
|
|
| Pages:
1
2
3
4
5 |








