| Pages:
1
..
3
4
5
6
7
..
10 |
prometheus1970
Hazard to Others

Posts: 138
Registered: 14-4-2010
Location: San Antonio, tx.
Member Is Offline
Mood: happy/ inquisitively eager
|
|
I'd really prefer to avoid any disonsety like the plague when pursuing this hobby. I fear that, should it come up, the fact that I lied
aboutsomething is more likely to make me look like a criminal (somebody with evil intent rather than just somebody who intends to break the law,
because it should not apply to those without ill intent). If my honesty precludes me from obtaining certain things, then I'll look to another primary
to synthesize.
Speaking of which, I just read Mr. Anonymous' thread on azo clathrates and he described that as an OTC synth l don't know of where to get azides for
that synthesis, so how is it OTC?
Just because you're paranoid doesn't mean everybody isn't out to get you.
|
|
|
hissingnoise
International Hazard
Posts: 3940
Registered: 26-12-2002
Member Is Offline
Mood: Pulverulescent!
|
|
Yeah, I know the feeling . . .
But if the honest approach drives you to experiment with unsafe materials then it'll be misplaced honesty!
http://www.readyreagents.co.uk/epages/es115347.sf/en_GB/?Obj...
http://www.readyreagents.co.uk/epages/es115347.sf/en_GB/?Obj...
|
|
|
markgollum
Hazard to Self
Posts: 53
Registered: 21-2-2004
Member Is Offline
Mood: No Mood
|
|
I have been reading about all the many things that people have been trying and proposing to get picramic acid.
I was once in a similar position, I had tried several experiments with ascorbic acid for example. I did not try the sodium hydroxide/sulfur reaction
since it had so many poor reviews at the E&W. Eventually I decided that I was going to do it right and use Na2S and that was that. The options
that I felt I had were as follows:
(1) Reduce sodium sulfate with carbon at 900-1000 C (could be done in steel plumbing pipe. If it is galvanized be sure to dissolve the Zinc with HCl
first).
(2) Reduce CaSO4 with carbon at 900-1100C then react the CaS with Na2CO3 in a solvent.
(3) Bubble H2S through NaOH in some solvent ( methanol comes to mind, you want to eliminate water as much as possible since it is virtually impossible
to properly dry the Na2S)
I ended up buying the Na2S before I tried the above options, and I must say that the classical sodium picramate synthesis is wonderful and produces a
beautiful, pure, free flowing product from which high quality DDNP can be made.
I eventually did try to produce Na2S via method 2 above. The carbon reduction went well but just takes time, an advantage is that any unconverted
gypsum is insoluble and can be easily filtered out with the excess/unreacted carbon. However, in the metatheses reaction with aqueous sodium carbonate
the sulphide solution is fairly rapidly oxidized by the air turning yellow. I attempted to get by this by just working quickly and while I was able to
get some hydrate of sodium sulphide, I did not titrate it. I tried to run the reaction in methanol and could not get is to go, despite running it
under magnetic stirring for a month.
I think that directly reducing sodium sulfate would be easy to do, it has the advantage of directly producing an anhydrous product that is easily
soluble in methanol allowing separation from reactants. (Anhydrous Na2S is quite soluble in methanol, Na2SO4 and hydrated Na2S are not)
Option 3 would only have been done as a last resort as the risks inherent with handling largish volumes of a fairly pure H2S gas stream are not to be
taken lightly.
[Edited on 24-1-2011 by markgollum]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
H2S is easy to produce either by acidifying a sulfide/polysulfide or possibly by heating paraffin and sulfur together...and this could be used as in
your scheme #3
or similar scheme. However, the sulfide route is one of many different methods which could work.
With regards to azides, sodium azide is itself producible via OTC materials.
The particulars for that have been described more than once in discussions
here and elsewhere. If I can search and locate those discussions for posting
a link here, then so can other people search and find. It has been an idea
of mine for a long time that some folks need to do a lot more reading before
they post to avoid covering things already covered before. Spoonfeeding gets old
and it just leads to more and more indulgence while the level of discussion
is not advanced. Reverse engineering has already been done, seek and ye shall find.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Many thanks for the ideas about producing sodium sulphide Markgollum! I will surely give the carbon reduction of gypsum a try one day. It is good to
know that producing (presumably) high purity picramic acid is possible using Na2S. The polysulphide reduction (NaOH and S) never gave good results,
either with homemade nitrite, (Lead+NaNO3 --> CO2) or reagent grade nitrite. The step to produce Na2S myself however was too much for me, given the
substantial danger of working with H2S and the uncertanty whether the picramic acid was the problem or the diazotization itself. 
[Edited on 24-1-2011 by nitro-genes]
|
|
|
markgollum
Hazard to Self
Posts: 53
Registered: 21-2-2004
Member Is Offline
Mood: No Mood
|
|
I would actually prefer to directly reduce sodium sulfate for the following reasons.
1) After extraction of fused product with methanol then boiling down the Na2S would be obtained in a fairly anhydrous form.
2) The reaction occurs in the liquid phase and would presumably be faster.
3) A lot more product could be produced.
One possible problem is that the carbon dust could float up to the top of the melt.
If this happens then it should be no problem to add so much carbon that the melt is more of a paste than than a liquid. The extra carbon should also
reduce the strength of the fused product making is easier to chip/hammer it out of the pipe.
One possible idea would be to melt and carbonize a mixture of sugar and Na2SO4 in a pan then use the black lumps in the pipe.
Of course you would want to prevent oxygen from the air from entering the pipe. One setup which I have applied in making calcium cyanamide (I haven't
characterized the cyanamide, but I basically followed the method Rosco has described: converting cyanuric acid to a sodium salt then precipitating the
calcium salt) in a pipe is in threading a short 6" section of steel air brake line to the top end cap. I stand the pipe up in my electric furnace
allowing the steel air brake line to protrude from a hole in the lid, this allows me to keep track of the reaction progress by lighting "flaring" the
evolving gasses (if excess cyanuric acid is used, it sublimes and can plug the tube so I keep a torch handy to heat the tube as well as a stiff steel
wire I can use to check for blockages).
|
|
|
papaya
National Hazard
Posts: 615
Registered: 4-4-2013
Member Is Offline
Mood: reactive
|
|
Thread seems old, anyway I want to ask if reduction of PA with oxalic acid will work yield picramate in boiling basic solution? This thought came to
me as I expect oxalic acid to decarboxylate on heating(basic solutions promote that, isn't it?) to give HCOOH which may be able to do the work. Sorry,
no real references can I provide, since searching on reductions with oxalic(or salts) gave nothing very interesting, except maybe unrelated this: http://pcat.cat.hokudai.ac.jp/document/pdf/NTB2009.pdf
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
I was under the impression that the reduction required a basic solution to prevent over reduction to triaminophenol, hence the neutralization to
picrates and scorbates that were discussed earlier. If so, then an oxalate salt would be required.
|
|
|
papaya
National Hazard
Posts: 615
Registered: 4-4-2013
Member Is Offline
Mood: reactive
|
|
Reading several papers it turns out that aromatic nitro reductions can yield different products depending on conditions and reducing agents. This
paper (final pages missing) http://www.sciencemadness.org/talk/files.php?pid=215569&...
"The Alkaline Reduction of Aromatic Nitro Compounds with Glucose" Though the paper starts with
"The fact that aromatic nitro compounds may be reduced by glucose in alkaline solution was apparently first discovered in 1865 by Braun who found that
on heating an alkaline glucose solution with picric acid a red color developed, which he believed was due to the formation of picramic acid."
it doesn't give any information if that was further confirmed, and their own findings indicate that glucose reduces nitro aromatics to azo- and azoxy-
compounds (coupling).
Same conclusions from book "FUNDAMENTAL PROCESSES OF
DYE CHEMISTRY", see chapter "reducion in general" it states "Reduction by glucose and alkali is used only in a very special field: the
reduction of nitro compounds to azoxy and azo compounds."
On the bright side, "THE REDUCTION OF SOME NITROBENZENES WITH ASCORBIC ACID"
http://dergiler.ankara.edu.tr/dergiler/31/1438/16170.pdf
it states that dinitrophenol is partially reduced to amino- nitrophenol with ascorbic acid, alkali and FeSO4 catalyst, I think that's going to work
also with PA.
What are your thought, especially on glucose - may it be that reaction with PA still yields picramic acid, regardless how differently glucose gives
azoxy- compound in other cases (though were with mono nitro), because sombody spoke here about blood(urine) sugar tests based on reaction with PA if I
remember correctly.
How do you people confirm that what you got is what you intended to get?
[Edited on 18-12-2013 by papaya]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I have been reading about DDNP for about 6 years now, off and on, and it has always seemed sort of illusive. Reading posts here and from the old Rogue
science forum it was clear that the big problem was obtaining a suitable reducing agent. The most common and effective selective reducing agents seem
to be sodium sulfide, sodium hydrosulfide and ammonium sulfide. After reading one of the excerpts from an early twentieth century chemistry book
posted on this forum, I decided to try generating hydrogen sulfide gas and bubble it through a sodium hydroxide solution in order to obtain sodium
sulfide or hydrosulfide. The method involves heating a mixture of low volatility hydrocarbon with sulfur. In this case Vaseline was used, but I have
seen examples where paraffin was specified. A pdf of the excerpt is also attached at the end of this post.
I have taken a lot of pictures, which should do most of the explaining. A Vaseline and sulfur mixture was put in a mason jar, which had a perfect hole
cut in its top to accept a rubber stopper. A piece of mild steel brake line was used to lead and cool the hot gas from the generator to the clear
vinyl tubing and bubbler at the other end. An aquarium air stone or bubbler was used as the diffuser.
Safety was of utmost importance, as very small amounts of this gas can kill you. It was a very cold (-20C) and breezy day. The generator was outside
and I was inside, behind a glass door that is reasonable airtight, controlling the power to the hotplate remotely with a variac (variable auto
transformer).
I increased the power to the hotplate gradually over the course of 30 minutes or so until a fairly steady, but slow, stream of bubbles was coming from
the diffuser. I didn't increase the power to the hotplate at this point because I believed slower production was safer. After an hour and a half I was
forced to stop as the sulfide/hydroxide solution started to freeze and the diffuser also froze which filled the backyard with H2S stink for 10-15
minutes. It was such a cold day, however, that no one was outside anywhere near where I was operating which was fortunate. Judging by the volume
increase at least 3/4 of the sodium hydroxide was converted to sulfide. According to Wiki the yellow colour of the solution is from polysulfide
contamination.
The last picture shows the reactants which were to be used for the following partial reduction producing sodium picramate. The sodium (hydro)sulfide
solution is on the left and picric acid made (days ago) from aspirin is on the right.
       
Attachment: Hydrogen Sulfide Gas Preparation_prothiere1903_abstract.pdf (100kB)
This file has been downloaded 862 times
[Edited on 27-1-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Next, a reduction of the ortho nitro group of picric acid, the one in the 2 position, was performed. Five grams of picric acid (from ASA) was
dissolved in a methanol-water solution and neutralized with sodium hydroxide. The solution was made from about 75% methanol and 25% water by volume.
I found a page with the reaction equations, going from TNP to DDNP, which was part of a Chinese primary explosives presentation. I have included a jpg
of that reaction equations page, as well as a link to the whole presentation.
http://www.cecd.umd.edu/documents/presentations/Hong-Kong/Yi...
The article I was reading when I performed the reduction is attached, and was also found on this forum. I believe Rosco posted it. I didn't follow it
accurately unfortunately, because of not knowing the process well and being distracted. The reaction time was much longer than recommended by the
article and it took me a long time to get the temperature up to where it should have been. I also was unsure of the concentration of my previously
prepared sulfide solution and I am sure I didn't use enough. A ferrous sulphate solution was used to test for reaction completion, but in retrospect
it was obvious that the endpoint hadn't been reached. I obtained about a 60% yield, and the article reported yields in some cases in the high 90's. I
still have at least 3/4 of my sulfide solution, so I will try again.
The colour change going through the reduction reaction is really very beautiful. The product in the last picture is (or should be) sodium picramate.
Conversion of the sodium picramate to picramic acid was easily accomplished by dissolving it in water and acidulating with sulfuric acid. Pictures of
the picramic acid produced, as well as the DDNP produced from it, will be shown in the DDNP thread.
        
Attachment: Picramic Acid using Sodium (Hydro)Sulfide J[1]. Chem. Soc., 1945, 663 - 665,.pdf (344kB)
This file has been downloaded 1578 times
[Edited on 27-1-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
"According to Wiki the yellow colour of the solution is from polysulfide contamination."
I've been told that sulfur contamination is detrimental, and you've gotten around that by going for H2S. Do you know what effect the polysulfides
would have on the final DDNP?
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I think the polysulfide contamination is only present in very small quantities. The yellow colour gradually developed after an hour and a half of
bubbling hydrogen sulfide into the sodium hydroxide solution. I believe a lot of lab grade solutions have a slight yellow tinge to them for the same
reason. If it was absolutely pure it would be clear, from what I have read.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
I can vouch for the yellow color of lab grade stuff. I have tech grade Na2S.3H2O and it forms slightly yellow solutions.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Actually after reading a bit more, it seems that sodium hydrosulfide is easily converted to polysulfides by oxygen. There could be more polysulfides
in that solution than I thought. There is a very good chance that I converted a lot of the sodium hydroxide all the way to sodium hydrosulfide.
From Wiki page on sodium sulfide:
"Sodium sulfide is the chemical compound with the formula Na2S, or more commonly its hydrate Na2S·9H2O. Both are colorless water-soluble salts that
give strongly alkaline solutions. When exposed to moist air, Na2S and its hydrates emit hydrogen sulfide, which smells like rotten eggs. Some
commercial samples are specified as Na2S·xH2O, where a weight percentage of Na2S is specified. Commonly available grades have around 60% Na2S by
weight, which means that x is around 3. Such technical grades of sodium sulfide have a yellow appearance owing to the presence of polysulfides. These
grades of sodium sulfide are marketed as 'sodium sulfide flakes'. Although the solid is yellow, solutions of it are colorless."
From Wiki page on sodium hydrosulfide:
"Solutions of HS− are sensitive to oxygen, converting mainly to polysulfides, indicated by the appearance of yellow."
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Just so it's clear, the green solution in the second picture above is slightly acidic aqueous ferrous sulphate. It was used as an indicator in order
to test for the presence of sulfide. A little of the reduction reaction mixture was extracted with a pipette from time to time and placed in a small
beaker, to which a little ferrous sulphate solution was added. A black precipitate of iron sulfide would show that the reduction was complete and that
there was unreacted sulfide in the reaction mixture. Obviously waiting a certain length of time after a sulfide addition before doing the test is
required.
Fe2(SO4)3 (aq) + 3 Na2S (aq) ---> 3 Na2SO4 (aq) + Fe2S3 (s)
The ferrous sulphate solution was prepared by dissolving fine steel wool in dilute sulfuric acid.
[Edited on 30-1-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
I was thinking just now, 'gee what about the excess NaOH in the reducing solution that you failed to neutralize, what will happen when it enters the
reduction reaction'. Its a good thing actually because the picric acid will over-reduce if done in an acidic solution. The sodium picrate is formed
to allow an alkaline solution for the reaction, using excess NaOH. I love it when a plan comes together.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I am pretty sure all, or most, of the NaOH was converted to at least sodium sulfide and probably mostly to sodium hydrosulfide actually. I didn't
actually test it, just judging by the quantities needed to reduce a give quantity of picric acid and the color of its solution, etc. Yeah, that was my
thought too, that a little extra NaOH in the reaction mixture wouldn't be a problem most likely.
[Edited on 30-1-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
Quote: Originally posted by Hennig Brand  | I am pretty sure all, or most, of the NaOH was converted to at least sodium sulfide and probably mostly to sodium hydrosulfide actually. I didn't
actually test it, just judging by the quantities needed to reduce a give quantity of picric acid and the color of its solution, etc. Yeah, that was my
thought too, that a little extra NaOH in the reaction mixture wouldn't be a problem most likely.
[Edited on 30-1-2014 by Hennig Brand] |
S-2, much like oxide does not properly exist in solution. In solution, it exists as equimolar HS- and OH-.
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Sodium Picramate - Experimental
Some more sodium picramate was made and this time greater care was used to record some of the reaction conditions and quantities used. This process is
not optimized and neither are the quantities used.
Five grams of picric acid (from ASA), of greater than 98% purity as indicated by melting point, was dissolved in 100 mL of methanol and heated to
50-55C. Sodium hydroxide (0.9g) was dissolved in 20 mL of water and with stirring was slowly poured into the alcoholic picric acid solution
neutralizing the picric acid and forming sodium picrate. While maintaining the temperature of the reaction mixture between 50-55C, sodium
(hydro)sulfide solution was pipetted in, with magnetic stirring, a mL at a time over the course of 20-30 minutes. The sodium (hydro) sulfide solution
was made by bubbling hydrogen sulfide gas through an aqueous solution of sodium hydroxide. Four grams of sodium hydroxide was used for the solution.
The amount of Vaseline and sulfur placed in the H2S generator was at least 50% more than would be needed if all H2S was absorbed and converted to
dissolved sulfide salt. The volume of sodium (hydro)sulfide solution obtained was ~45mL. About 15 mL was used to reduce the 5g of sodium picrate to
sodium picramate. Ferrous sulfide solution was used to test for reduction reaction completion. After the last sulfide addition, heating was maintained
for an additional 10-15 minutes and then the heating was stopped and the reaction mixture allowed to cool. Once the reaction mixture had cooled to
about 40C, about 75 mL of ice cold water was added and the beaker was set into an ice-water bath and cooled very quickly to 10C or below. The sodium
picramate was then filtered out and washed with small quantities of ice cold water.
From 5g of picric acid, 3.8g of sodium picramate was obtained or about a 79% yield. My last experiment produced only a 60% yield. The biggest
difference between the two experiments was that the first experiment used less methanol and more water as the reaction solvent/medium. A little
greater care was also used when testing for reaction completion, though I am not sure I really got it right yet. There is a lag time, governed by the
reaction time, between when the reducing agent is added and when it is consumed.
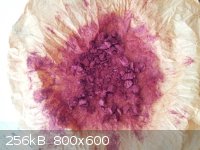
[Edited on 4-2-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The absence of any sulfur as a trace impurity would be desirable, particulaly for a DDNP destined to be mixed with chlorate since sulfur and chlorate
are a bad storage stability, known incompatability. This is one reason why alternative means of reduction were examined.
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
Are you implying that the sodium(hydrogen)sulfide isn't appropriate for this reduction? It seems to isolate the sulfur from the reaction.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I think he is referring to some of the non sulfide reducing agents that have been experimented with. The mixed polysulfide reducing material (made the
cookbook ways from sodium hydroxide and sulfur) is apparently bad for producing a lot of sulfur contamination. I tried the cookbook method a few years
back, but it didn't work for me. I don't think that sulfur contamination would be much of a concern with the sodium (hydro) sulfide solution of
reasonable purity. My H2S gas generator and absorption setup is a joke though. It does work, but the gas could be cleaned before it got to the bubbler
and a lot could be done to improve safety and improve absorption of the gas.
I have made enough chlorate/sulfur/aluminum crackers to know that chlorate and sulfur are not at all storage stable. Small amounts decomposing in
storage don't ignite or anything (that I have seen), they just degrade in a matter of weeks to a state where performance is severely diminished.
[Edited on 5-2-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
dave321
Harmless
Posts: 45
Registered: 22-11-2012
Member Is Offline
Mood: No Mood
|
|
reduction of styphnic acid with tin chloride
on a similar note to the reduction of picric acid, why in the following patent would the use of tin chloride be preferred to the sodium sulphide
approach used for picric acid ?
Attachment: USE OF TIN CHLRIDE FOR REDUCTION.pdf (240kB)
This file has been downloaded 879 times
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
There is an inevitable sulfur impurity and diaminonitrophenol impurity in the picramic acid produced by a sulfide reduction. That is why purification
schemes have been used for the picramic acid to free it from such impurities. Other reduction schemes for producing picramic acid from ammonium
picrate or sodium picrate using other reducing agents are probably superior in terms of avoiding the impurities. Many reducing schemes not involving
sulfides can likely be used so long as the reduction is carried out at a temperature and pH that favors the production of picramate, meaning mild
temperatures and alkaline reaction systems.
Alternate possible reduction schemes would include sodium ascorbate, ferrous sulfate, manganous sulfate, sodium bisulfite, zinc powder or zinc
amalgam, aluminum amalgam, iron filings, magnesium turnings, possibly formaldehyde or hydrazine, or electrolytic reduction.
The hydroxy DDNP analogue of US4246052 attached again here and its salt forming ability looks interesting and promising. The hydroxy DDNP may be
subject to forming azide and hydrazine derivatives similarly as does DDNP which could increase its energy even further. Interesting how the hydroxy
picramic acid may be obtained from a reduction of styphnic acid in acidic medium, because it may follow the same track and be more easily reducible in
alkaline medium, as does occur for the reduction of a picrate to a picramate.
Attachment: GB412460 Lead Dintrophenylazide.pdf (331kB)
This file has been downloaded 869 times
Attachment: GB406228 Lead Dintrophenylazide.pdf (172kB)
This file has been downloaded 867 times
Attachment: US2728760 Kenney , hydrazine derivative lead salt of DDNP.pdf (124kB)
This file has been downloaded 798 times
Attachment: US4246052 SnCl2 reduction of Styphnic Acid and DDNP analogue therefrom.pdf (240kB)
This file has been downloaded 813 times
[Edited on 5-2-2014 by Rosco Bodine]
|
|
|
| Pages:
1
..
3
4
5
6
7
..
10 |








