| Pages:
1
..
22
23
24
25
26
..
33 |
nitro-genes
International Hazard

Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Another random crazy idea with regard to OTC production of styphnic acid and DDNR:
Would it be possible to convert picric acid to styphnic acid using fenton chemistry? Phenol can be synthesized from benzene by ferrous sulfate and
hydrogenperoxide, generating the extremely reactive hydroxyl radical, which is able to replace an aryl hydrogen. Another example is supposed
dinitrocatechol intermediate in destruction of dinitrophenol using fentons reagent. Reaction with 2 ortho nitrogroups unknown, possibly replacement of
nitrogroup? Since percarbonates are OTC, the idea might not be worth a try.. 
[Edited on 9-11-2015 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks to Solo for uploading the article!
Seems Klapotke's group has done a folow up on the dinitrodiazophenols, summerized in a recently published paper (attachment). The article has some
interesting features, though the exact scope of the article is not completely clear, the title suggests testing of the initiation capabilities of some
of the novel diazo compounds described, whereas the most promominent feature of the article is mainly the nitration of N,N protected
p-phenylendiamines using different nitration and deprotection schemes. Maybe they were looking for an efficient 1 or 2 step synthesis for the DDNR or
the tetranitro derivative of the p-diamine as they claimed, though could not be realized this way. The nitration of p-chloroaniline seemed like a good
candidate for the former, too bad the chlorine won't budge. An interesting features is also the first time shown existence of 3-amino DDNP, presumably
from the mobile nitrogoup rerangement to the nitramine and decomposition to the diazo during the deprotection in sulfuric acid. It has expected very
high density and roughly a DDNP comparable sensitivity to impact. Some flame sensitivity test video clips in the supplementary should be obligatory, I
mean, isn't that why we make these things? 
The HODDNP (DDNR sound better IMO, but maybe has a to SMDB'ish ring to it )
really seems a very potent initiator, not only was 10 mg was enough to initate RDX, the DDNR itself seems very stable to heat and moisture (for a
diazo compound anyway), although acidity and metal compatibility may still be an issue. For that matter, it may also be nice to see properties of some
salts of DDNR.
Personally would have liked it if they had also included a more thorough analysis and comparison between o-DDNP and p-DDNP, maybe using PETN for more
resolution between the two and some statistics, but then again the initiating difference between the two likely is not that large anyway, so more of
an academic discussion than of real practical value.
Attachment: Synthesis and Initiation Capabilities of Energetic Diazodinitrophenols.pdf (754kB)
This file has been downloaded 990 times
[Edited on 7-12-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
There is a conspicuous absence of original references and history regarding the DDNR and an odd avoidance of calling DDNR what it is and strangely
applying a new naming to an old compound.
Anyway it seems they are going to some trouble to ignore information already presented here in our discussion and avoid questions raised also.
It just seems very odd, because we know that they are aware of our discussion here.
We have already identified simpler routes to DDNR via the original work of Benedikt and Hubl from mononitroaminoresorcinol or dinitroaminoresorcinol
and other schemes from resorcinol or styphnic acid as the starting material.
We have also identified the route for DDNR via the further acetylation of paracetamol using acetic anhydride to form the diacetyl derivative which may
be nitrated to the trinitro compound, and deacetylated,
and diazotized with a concurrent loss of the 3-nitro decomposed to a hydroxyl to form DDNR, similarly as reported in the earlier Klapotke article, and
earlier by Meldola who is still not credted by Klapotke.
It seems they are going out of their way to not identify the actual history of DDNR.
And it is still an unresolved issue that the compound is a 6-diazo as Klapotke is identifying DDNR, which would require a diazo rearrangement from the
4-diazo as would initially form from a 4-amino precursor, and this seems unlikely. I think this follows somewhat the earlier confusion of Meldola
about a supposedly "mobile" nitro group that later turned out to be not mobile at all

[Edited on 12/7/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
The naming is odd, I propose DiDiDIB or BeDiDIDI instead (dihydroxydinitrodiazobenzene), which sounds better than HODDNP.
Why the original historic references are not included is also surprising, perhaps it is as simple as that citing articles from the 1800's just doesn't
seem like cutting edge science, many journals actually prefer not to include "out dated" references. The DDNR seems like a really interesting
initiator though and a cheap, safe, up scalable synthesis of the DDNR is potentially very lucrative. A problem with the older described synthesis
routes may be that the use of salts in combination with the DDNR may pose safety issues on an industrial synthesis scale, as the potassium salt of
DDNR reportedly exploded on recrystallization. On the other hand, how valuable would the DDNR really be, except maybe in certain stab detonator type
designs? Especially in a time where much more heat stable and brisant high nitrogen primaries are continuously being developed.
Regarding the formation of compound 4 from 2; mobility may not be the word here, I meant that hydrolysis of one of the two adjacent nitro groups will
slowly form nitric acid with the small amount of water present in the sulfuric acid used for deprotection, which may rearrange to a more stable
nitramine , upon dilution with water this would decompose further to the diazo. I observed something similar when I tried to nitrate the isopicramic
further using sulfuric acid and 2 mole eqv of KNO3. Upon crashing in ice, initially a bright yellow substance formed that upon drying for a long time
formed a more orange coloured substance that I was positive to be p-DDNP, the most likely explanation is that the initial bright yellow stuff was the
nitramine.
Alternatively, the process may involve some intramolecular redox/rearangement similar to bamberger, involving partial reduction of the nitrogroup and
formation of the quinone first. When I drew the reaction equation for the diazotization of isopicramic I already wondered about this possibility,
though would think that the presence of the other nitrogroups would prevent this from happening. Another possibility is that compound 4 may have originated from a reduction by the solvent used, this wouldn't happen
this way though in a totally inert organic solvent.
[Edited on 8-12-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I was thinking about the disputed "rearranged" 6 diazo of DDNR from the earlier Klapotke article reiterated in this present article called 6 diazo
again, which I believe is more probably a 4 diazo.
It was something I brought up earlier several times as a structure disagreement and I am still of the same opinion as stated earlier
in the post linked and other posts earlier and later also. NMR notwithstanding it would seem it has to be a 4-diazo for DDNR or else there is a
bizarre diazo mobility and migration from 4 to 6. Meldola has to be laughing from the great beyond, oh no not another mobile substituent group, first
the magic? nitro and now the magic? diazo just migrating across the ring...you got to be kidding me.
http://www.sciencemadness.org/talk/viewthread.php?tid=439&am...
We are having too much fun to be unpaid for the good effort. Maybe a fat government defense contractor grant and suddenly all the NMR position 6 diazo
library data is put in jeopardy or patent position is at risk, if the "new" compound is really an old compound DDNR already at least twice patented
before and published even long before that.
Tom Klapotke et al should not throw all that government grant money away, just do a wire transfer to Rosco and Nitro and we will soon get to the
bottom of this puzzling matter. Or maybe we could go on a chemical safari.
Have Beaker Will Travel Will Work for Beer and Women
Shucks too late ....Benedikt and Hubl already published in May 1881 which was 134 years ago
I would not risk being called a gender insensitive "pimp chemist"
for having too much HODDNP and CUNT in the laboratory and I would expect that some of the female persuasion coworkers might suggest a different
nomenclature  
[Edited on 12/8/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Maybe we can try crowdfunding, small donations earn an on demand designed explosive molecule, a really large donation and you can build your own
intercontinental missile or something (Haven't worked the details out completely yet). Would be really nice to legaly buy things like 100% nitric,
instead of distilling it everytime from draincleaner.
Seriously though, these nitration schemes are pretty interesting, although seem to largely behave as expected from previous literature. The formation
of compound 8 seems to resemble that as described for the dinitro o-toluidine "J. Org. Chem. 1986,51, 2572-2578: Synthesis of Polynitrodiazophenols",
although the mechanism still eludes me. Seems I was wrong about the hdyrolysis, since here they used DCM and evaporation. Different for the
p-chloroaniline is the presumable presence of 2 ortho nitro's (respectively to the aminogroup) after nitration, very interesting. But if this would be
the case, the diazo be unlikely to form (similar to TNA or picramide). Really seems the formation of the trinitrochloroaniline or 2,6-dinitro
3-hydroxy 4-chloroaniline would be more favorable. Presumably, the 3-nitro and/or 4-chloro make that the 2-nitrogroup and aniline (or nitramine)
interact to form some sort of unstable intermediate, that decomposes to leave the diazo. This would also be quite different however from the nitration
of 3 nitro-aniline or hydrolysis of the resulting tetranitroaniline, which produces only 3-aminopicric and no diazophenols. ...puzzling...Maybe Philou
can shed some light on this matter.
I would still be curious for the p-iodo or bromoaniline, or perhaps a carboxyl group somewhere could help.
Also interesting that compound 2a (the trinitrophenylenediamine) only forms from high water content nitric. I suspected that the protected amino might
still protonate and pose deactivating and meta direction to the ring, though maybe it prevents formation of the secondary nitramine, which might also
be deactivating or lead to decomposition. Apart from the better hydrolysis resistance, maybe the bulkyness of the methylsulfo protection group used
also helps here? Also wondered if this is the reason why acetaminophen can't be nitrated to the trinitro, as it is simply too prone to oxidize from
65% nitric alone, maybe very low temperatures and a very specific water content could also do.
Very interesting point in the article is also that the heat test of DDNR (5 mg) didn't lead to detonation. I'm curious now if the isopicramic/65%
nitric acid diazotization didn't produce some DDNR afterall, since the "explodes on heating" was the only real real differentiation point I had for
distinguishing the DDNR from the DDNP. Colour means little, since it depends on many things like crystal shape and impurities formed.
[Edited on 10-12-2015 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
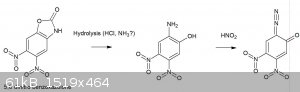
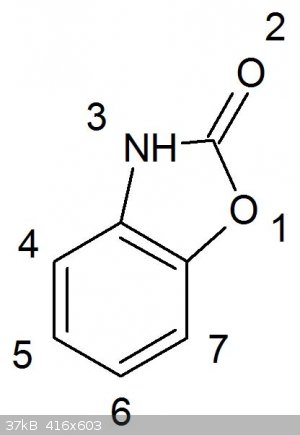
This was briefly mentioned before in the DDNP thread, though now I’ve had the time to do some reading regarding the potential use of benzoxazolone
to produce 2-diazophenols. In general, ring opening and deprotection are problematic and I doubt the nitration of benzoxazolone is able to produce di
or trinitro isomers other than the described ones below.
Benzoxazolone can be prepared by numerous routes, of which some are easily in reach for the amateur, completely OTC and high yielding:
1. Reaction of 2-aminophenol and urea in a mineral acid solution (sulfuric acid monohydrate, HCl, phosphoric US3812138)
2. Hofmann rearrangement of salicylamide under strongly basic conditions (CA 1150276 A1)
In general, the 6 (meta to the hydroxyl) and 5-positions of benzoxazolone are most activated, accordingly nitration using 65% nitric acid produces
almost exclusively 6-nitrobenzoxazolone, while further nitration using fuming nitric acid gives 5,6 dinitro benzoxazolone [1].
Although nitration using nitric acid alone is well described in literature, I couldn’t find many references regarding the products using mixed acid
nitration (nitric/nitrate salts+sulfuric). One option could be that concentrated sulphuric acid (being a stronger acid) leads to opening of the
oxazolone ring, which could influence the orientation during nitration to produce different isomers than those already described. Then again, heating
of 2-hydroxy phenylurea in sulphuric acid is mentioned to lead to ring closure and patent US3812138 describes condensation of urea with 2-aminophenol
using sulphuric acid to yield benzoxazolone, which would imply that either this depends on pH /sulphuric concentration or benzoxazolone is reasonably
stable in concentrated sulphuric, but not soluble enough or too activated for mixed acid nitrations.
One reference in which mixed acid nitration of benzoxazolone to 6-nitro benzoxazolone (92% yield) is mentioned is “Synthesis of
2-Amino-5-Nitrophenol by Two Step Process, Yin Zhenyan and Li Yanyun (2007)” They further describe hydrolysis the 6-nitro benzoxazolone using 96%
ethanol and NaOH, which would represent a usable OTC route to 2-amino 5-nitrophenol.
Anyway, an idea that I had was that perhaps starting with 65% nitric and gradual addition of sulfuric acid (or even AN/SA and a good icebath) may
produce 5,6 dinitro benzoxazolone directly. Would it be possible to hydrolyse the oxazolone ring without hydrolysis of one of the 2 nitro groups? If
this would give 2-amino 4,5-dinitrophenol and diazotized would this lead to an undescribed isomer of DDNP, or would it be unstable? Reportedly,
2-diazo 5-nitrophenol is pretty stable and used in coupling reactions for dye manufacture. My hopes for the hydrolysis of the 5,6 dinitrobenzoxazolone
are not too high though, due to the reported stabilizing effect of a 5-nitro group against hydrolysis of the oxazolone ring [2] in combination with
likely increased susceptibility towards hydrolysis of the 2 adjacent nitro groups.
Anyway, any input on the behaviour of benzoxazolone during mixed acid nitrations or other ideas regarding the synthesis of energetic derivatives from
benzoxazolone would be appreciated.
[1] Synthesis of Some Substituted Benzoxazolones
Robert L. Clark, Arsenio A. Pessolano
J. Am. Chem. Soc.
1958, 80 (7), pp 1662–1664
DOI: 10.1021/ja01540a038
[2] Benzazole, IX. Über das chemische Verhalten der Nitro-benzoxazolone
HELMUt Zinner, Herbert HERBIG
European journal of inorganic chemistry
Februar 1959,Volume 92, Issue 2 Pages 407–414
DOI: 10.1002/cber.19590920224
[Edited on 19-9-2016 by nitro-genes]
Attachment: phpLqOh08 (470kB)
This file has been downloaded 612 times
Attachment: Synthesis of Some Substituted Benzoxazolones.pdf (350kB)
This file has been downloaded 680 times
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Also interesting:
This article describes the synthesis of a p-diazo-o-nitrophenol (presumably 4-diazo, 2-nitrophenol, but no detailed chemical analysis is performed),
which seems chemically incredibly stable and maybe faster accelerating than diazodinitrophenols, as it reportedly exploded with a very loud bang and
display of high brisance during an ignition temperature test. Should be doable from acetaminophenol, though probably much more sensitive than p-DDNP.
"Das o- N i t ro-p-d i a zo i urn p h en ol explodiert scharf bei 168 C. unter starkem Knall, wobei in der Regel das Schwefelsaurebad, in dem
der Explosionspunkt bestilnnit wurde, in Triimmer ging. Ebenso tritt Explosion durch Schlag ein. Es ist in konzentrierter Schwefelsaure und sogar in
konzentrierter, rauchender Salpetersaure glatt 10slich und scheidet sich, wenn auch nicht ganz quantitativ, beim Verdiinnen rnit Wasser a n v e r a n
d e r t wieder ab. Konzentrierte Jodwasserstoflsaure (1.7 g) bewirkte unter starker Erwarmung Stickstoff-Entwicklung. In heissem Wasser ist das
o.Nitro-1?-diazoniumphenol loslich, langeres Kochen bewirkt unter Stickstoff-Entwicklung Zersetzung. Es scheidet sich dabei eine braune, vollkolnnien
amorpbe Masse ab, die beim Erhitzen am Platinblech verpufft. Von den organischen Liisungsmitteln ist das o-Nitro p,-diazoniumphenol nur in siedendem
Aceton und Alkohol lijslich, vollkommen unloslich ist es in Ather und Benzol. Bei 100° l a s t es sich tagelang aufbewahren, ohne an Gewicht
ahzunehmen. Gegen Licht scheinen die gelben Bliittchen etwas empfindlich zu sein, da sie sich nach einiger Zeit stark braun farben. Da von vorherein
eine Verbrennung des o-Nitro-p-diazoniumphenolsw egen der ungemein starken explosiven Eigenschatt unausfiihrbar war, wurde die Substanz nur auf ihren
Stickstoffgehalt hin analysiert"
[Edited on 20-9-2016 by nitro-genes]
Attachment: Uber die Bildung des o-Nitro-p-diazonium-phenols..pdf (474kB)
This file has been downloaded 641 times
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
2-nitro 4-diazophenol is nowhere nearly as energetic as described in the article above, which is no surprise. Diazotization of the 3-nitro
4-methoxyaniline producing the nitrosamine probably rearranges (unlike the unalkylated phenol) directly to a 2-nitro-3-hydroxy-4-diazo-1-anisole
(maybe via a cyclic intermediate), which is easily nitrated further (like von Herz has described upon boiling with nitrite) to produce DDNR. This is
really interesting, with regard to OTC produced DDNR from acetaminophen it is also interesting how 2-nitro-4 aminophenol would behave when diazotized
using 65% nitric, the order of nitro group additions is not really clear yet. Also, it may be possible that diazotization followed by nitration or
maybe even direct nitration of methoxy 4-aniline (or other O-alkyated 4-anilines) or 2-nitro 4-methoxyaniline using 65-100% nitric acid or
combinations of sulfuric acid and nitric acid may produce DDNR in one reaction.
[Edited on 23-9-2016 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Mono nitration of acetaminophen:
5 grams purified acetaminophenol was finely powdered and added to 16 grams 97% sulphuric acid at 0 deg C. until everything dissolved. In a separate
beaker 2.7 grams of dry ammonium nitrate was dissolved in 8 g 97% sulphuric acid and. While keeping temperature between -10 and -5 (to favour
mononitration, for as far as that is possible), the AN/SA was added dropwise to the acetaminophen/SA solution. Total addition took 1 hour, and was
left to stir for an additional 15 minutes on ice. The clear yellow solution was added to 60 grams of crushed ice and stirred vigorously. Some sticky
orange-yellow stuff separated and was removed from the beaker. The rest of the clear yellow solution was left in the fridge overnight upon which an
estimated 2 grams of a bright yellow crystalline substance had separated (presumably 2-nitro 4-acetamidophenol). To the 2 grams of the yellow
substance, 4 grams sulphuric and 4 grams water was added, and kept at reflux for 2 hours, creating a yellow solution. About 1 ml of the solution was
added to a separate beaker and 5 grams of crushed ice added. This precipitated a light yellow micro crystalline substance, presumably the diazonium
sulphate salt. This was diazotized at 0 deg C for 30 minutes using sodium nitrite, the light yellow solid filtered washed and dried for 24 hours.
Additional drying for 30 minutes at 100 deg C improved burn rate significantly. It’s pretty energetic for something so oxygen defficient.
Further nitration in 65% nitric could possibly yield some DDNR, though, also the diazonium nitrate is likely meta directing resulting in p-DDNP again,
but who knows. 2-nitro-4-diazophenol, might also form nitrate or perchlorate
salts
Attachment: 2-nitro 4-diazoniumphenol - Copy.avi (4.2MB)
This file has been downloaded 1016 times
I think there is no completely OTC way of producing DDNR (without O-alkylation) using only acetaminophen and nitric/sulfuric, although I would still
like to try nitration of 2-nitro-4-diazoniumnitrate and 2-nitro-4-aminophenol and possibly 2-amino-5-nitro-phenol from the hydrolysis of
6-nitrobenzoxazolone.
[Edited on 24-9-2016 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
I keep talking to myself, but ok, here it is anyway
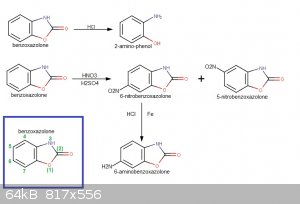
Still had some benzoxazolone left from the synthesis described by Nlegaux (https://www.sciencemadness.org/whisper/viewthread.php?tid=63...), so I decided to try the nitration/reduction to 6-amino benzoxazolone. The
benzoxazolone ater recrystallization from ethanol/water (cream colored needles) melts at around 140 ish deg C (literature, 138C). When together with
salicylamide on the hotplate, the benzoxazolone melts sharply right before a clean sample of salicylamide melts (literature, 142 C).
Nitration of benzoxazolone:
10 grams of 65% nitric and 5 grams of 96% sulfuric acid was added to a 20 ml beaker and cooled to 0 deg C. using an icebath. In small additions, 2
grams of benzoxazolone was added over about 30 minutes. This was kept stirring on ice for an hour and then allowed to stir at room temperature for 15
minutes or so, the mixture had thickened considerably at this point and was poured on ice. After drying at room temperature for a week, this gave 2.1
grams of cream coloured putative 6-nitrobenzoxazolone. When on the hotplate, it starts to blacken at around 200C, melting at 240C. With the hotplate
at 250C a sample melts instantly (literature 240C). Both melting point and yield of the 6-nitrobenzoxazolone is much higher using the nitric/sulfuric
acid nitration instead of using 65% nitric at 50C, suggesting that either less oxidation is occuring this way, or this nitration scheme is more
selective towards 6-nitro instead of the much lower melting 5-nitro isomer.
Reduction of 6-benzoxazolone to 6-amino benzoxazolone:
The reduction of 6-nitro benzoxazolone was performed as described in some article I found. To 70 ml water was added 2 ml of 8% HCl and brought to 90C.
Then 6 grams of steelwool was added in slightly compacted chunks. Over the course of 30 minutes, 2.1 grams of 6-nitrobenzoxazolone was added in small
portions. The dissapearance of yellow colour is a good indication that complete reduction has occured, which only took about 45 minutes. Then 2.5
grams of NaOH in 10 ml water were added, everything heated to 50 deg and the iron oxides filtered off. The basified filtrate containing the sodium
salt of 6-aminobenzoxazolone turned slighly reddish over the course of several hours. The filtrate was neutralized with dilute HCl to precipitate the
6-amino benzoxaolone. This needs to be done carfully with good lighting to precipitate most of the product. If the pH is lowered little too much,
everything goes immediately in solution again. Final yield was 1.2 grams of brownish/white glittering crystals that melted sharply at around 200C
(literature 202C), brownish colour probably from sligth oxidation or iron contamination. Product was not recrystallized.
Nitration of 6-aminobenzoxazolone:
Expecting that 6-aminobenzoxazolone is very susceptible to oxidation, the nitration was attempted on a very small scale.
Using 65% nitric:
3 ml's of nitric acid was added to a 10 ml beaker and cooled on ice, about 100 mg of's of 6-amino benzoxazolone was added in small portions. Upon
contacting the nitric, the crystals seemed to blacken but no gas was evolved and slowly started to dissolve. After stirring for about 15 minutes the
solution had turned a dark red but nothing further seemed to happen and the temperature was raised to 20 deg C upon wich the solution turned a dark
violet colour in a about 30 minutes with no evolution of gas, then the temperature was raised further to 50 deg C, which produced a darker solution,
but still no evolution of gas. Out of curiosity, a small crum of sodium nitrite was added, some NOx was evolved and kept slowly evolving until the
mixture had taken on a light orange colour. Ice was added, but nothing precipitated, neutralization and salting out produced no precipitate either.
suggesting that most of the product was likely destroyed.
Using AN/SA:
4 grams of AN was added to 16 grams of 96% SA and cooled to 0C using an icebath. Small additions of the putative 6-aminobenzoxazolone (500 mg) were
made over the course of about 30 minutes. Upon contacting the mixed acids, there was no gas evolution. Most of the crystals dissolved very quickly
after a small delay (with some dark specks remaing, maybe iron contaminants), turning the solution an orange to a dark reddish/orange after all
additions had been made. No gas was evolved at this point. About 15 minutes after the final addition the mixture was still a dark reddish/orange,
though very suddenly gas evolution became evident with many small bubbles being visible in the mix. Stirring was continued for several hours, during
which gas evolution remained evident and constant. When no gas formation could be observed anymore (5 hours), the mixture was drowned in ice, no
precipitate resulted.
It seems that the 6-amino group of 6-aminobenzoxazolone is very susceptible to oxidation, probably due to the stabilization of the imine intermediate
due to the lactam-lactim resonance of the oxazolone group. Would still be interesting to try and acetylate the 3 and 6 amino groups, but this needs
acetic anhydride again. It is still strange how this could happen under very acidic conditions using mixed acids though, since I would have guessed
this would deactive both amines and introduce a 5-nitro, leading to further nitration. Maybe I should have recrystallized the 6-amino benzoxazolone
futher, the purple colour is likely some iron complex.
[Edited on 16-11-2016 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Not speaking to yourself...speaking to all of us ;-)
"Reduction of 6-nitro-benzoxazolone to 6-amino benzoxazolone:"
The red part is missing.
I also have 1000 ideas involving diazoniums but no personal time or space to work for it (ex-girlfriend stil living at home while she should have left
in march 2016  ). ).
When she had left, I will be able to experiment more and repeat some of your experiments and eventually pay you a visit (or you come to my place).
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks Philou, would be interesting to hear some of your ideas regarding the synthesis of novel diazophenols.. For me personally, trying to synthesize
some of the diazophenols from relatively unstudied and OTC starting materials gives me some spare time relief. I'm not necessarily looking for a
specific product, just enjoying the chemistry and lessons learned from them. So much possibilities regarding these nitrations, hydrolysable nitro
groups, internal rearrangments of all kinds, mobile halogens and potentially explosive compounds...chemistry at it's best.  Benzoxazolone seemed interesting, one of the things I would still like to do is
the NA/SA mediated nitration of 6-chlorobenzoxazolone, whether nitration would stop at 5-nitro-6-chloro, or perhaps a second nitro group may be
introduced due to the less deactivating chloro group. Kind of worried this may also produce some dioxin like structures though. Benzoxazolone seemed interesting, one of the things I would still like to do is
the NA/SA mediated nitration of 6-chlorobenzoxazolone, whether nitration would stop at 5-nitro-6-chloro, or perhaps a second nitro group may be
introduced due to the less deactivating chloro group. Kind of worried this may also produce some dioxin like structures though.
[Edited on 17-11-2016 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
This may be interesting for you...6-carbamido-benzoxazolone from 6-amino-benzoxazolone and K cyanate.US Patent 2806853 A (downloadable into pdf format from there by clicking "téléchargez le pdf")
So
H2N-C6H3(-O-CO-NH-)cyclo turns into H2N-CO-NH-C6H3(-O-CO-NH-)cyclo
This may help prevent oxydation when nitrating.
To help a bit (by fixing the idea) I have made a synthetic pathway of your reaction...a drawing speaks more than a text...at least it helps me to have
a visualization to refer to.
[Edited on 18-11-2016 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Quote: Originally posted by nitro-genes  |
Nitration of 6-aminobenzoxazolone:
Expecting that 6-aminobenzoxazolone is very susceptible to oxidation, the nitration was attempted on a very small scale.
Using 65% nitric:
3 ml's of nitric acid was added to a 10 ml beaker and cooled on ice, about 100 mg of's of 6-amino benzoxazolone was added in small portions. Upon
contacting the nitric, the crystals seemed to blacken but no gas was evolved and slowly started to dissolve. After stirring for about 15 minutes the
solution had turned a dark red but nothing further seemed to happen and the temperature was raised to 20 deg C upon wich the solution turned a dark
violet colour in a about 30 minutes with no evolution of gas, then the temperature was raised further to 50 deg C, which produced a darker solution,
but still no evolution of gas. Out of curiosity, a small crum of sodium nitrite was added, some NOx was evolved and kept slowly evolving until the
mixture had taken on a light orange colour. Ice was added, but nothing precipitated, neutralization and salting out produced no precipitate either.
suggesting that most of the product was likely destroyed.
Using AN/SA:
4 grams of AN was added to 16 grams of 96% SA and cooled to 0C using an icebath. Small additions of the putative 6-aminobenzoxazolone (500 mg) were
made over the course of about 30 minutes. Upon contacting the mixed acids, there was no gas evolution. Most of the crystals dissolved very quickly
after a small delay (with some dark specks remaing, maybe iron contaminants), turning the solution an orange to a dark reddish/orange after all
additions had been made. No gas was evolved at this point. About 15 minutes after the final addition the mixture was still a dark reddish/orange,
though very suddenly gas evolution became evident with many small bubbles being visible in the mix. Stirring was continued for several hours, during
which gas evolution remained evident and constant. When no gas formation could be observed anymore (5 hours), the mixture was drowned in ice, no
precipitate resulted.
|
You are facing several problems/challenges here!
1°) ortho and para compounds of the type amino/hydroxy are very succeptible to oxydation (formation of o- (or p-) quinon) sometimes leading to black
polymerized tars...complete decay of the molecule into CO2 is rare.
2°) not strong enough HNO3 may lead to diazotation (by self catalytic decomposition into HNO2 while oxydising part of the substrate) and into this
case to a triazole p-quinon-imine bridge between the H2N- (diazotized into HO-N=N-) and the available acidic H form the Acyl-NH-..
3°) The prioritization of introduction of -NO2 in ortho or para position
HO- > H2N- > Acyl-NH- > Acyl-O- (> = stronger than)
Here by introducing an extra NH2- it becomes collaborative with the Acyl-O- and so they will favor entrance of the -NO2 into position 5 or 7 (or both
since the new introduced -NO2 favors meta vs itself) but this is true only if the oxazole ring survives the first or second NO2 introduction because
otherwise you are playing eather with 2,5-diamino-4-nitro-phenol (or/and 2,5-diamino-6-nitro-phenol) or with 2,5-diamino-4,6-dinitro-phenol.
Such compounds will probably oxydise and split off a nitro for an HO- (see nitranilate (2,5-dihydroxy-3,6-dinitro-paraquinone) formation from
chloranile and NaNO2).
So quite complex!
Into your first trial, I'm not sure you got nothing...maybe the compound was too soluble...
--> an extraction with an organic solvent and evaporation would have told you something was there.
--> or maybe the introduction of a heavy metal to precipitate a salt (like AgNO3, Hg(NO3)2 or Pb(NO3)2 (this is not applicable to your second test
because sulfates of Hg(2+), Pb(2+) or Ag(+) would precipitate)
About your quest for Diazodinitroresorcinol (DADNR),
I think you are on a good track...
Chlorination of 6-aminobenzoxazole via diazotation is a good idea... but I would go straight from 6-nitrobenzoxazole.
1°) hydrolyse the nitrobenzoxazole with HCl to get 2-amino-5-nitrophenol
2°) nitrate further since hydroxy will prevail it will introduce a second nitro into position 4 or 6 (or both)
3°) then it will hydrolyse the NO2 into position 5 and at the same time diazotise to o-DADNR
Both routes are prompt to easy oxydation because amino-phenol in ortho vs each other.
Eventually diazotise directly 2-amino-5-nitrophenol into 5-nitro-ortho-diazophenol and nitrate futher to 4,5,6-trinitro-orthodiazophenol (diazo is
indeed a meta director) and submit to water for hydrolyse of the 5-nitro to DADNR
But this route involve the use of explosive diazo compound from the begin to the end and increase of the OB with increasing amount of NO2 -->
Caution, low heat/agitation, an tiny minute amounts.
Working into a dillution solvent unreactive towards HNO3 might be a must for safety (CH2Cl2/conc HNO3 for example)
Last idea...probably safer:
work from meta chlorophenol, trinitrate to 2,4,6-trinitro-3-chlorophenol, hydrolyse to TNResorcinol and submit to NH4SH for a mono nitro group
reduction...then diazotize to o-DADNR or p-DADNR.
[Edited on 18-11-2016 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by nitro-genes |
Nitration of benzoxazolone:
10 grams of 65% nitric and 5 grams of 96% sulfuric acid was added to a 20 ml beaker and cooled to 0 deg C. using an icebath. In small additions, 2
grams of benzoxazolone was added over about 30 minutes. This was kept stirring on ice for an hour and then allowed to stir at room temperature for 15
minutes or so, the mixture had thickened considerably at this point and was poured on ice. After drying at room temperature for a week, this gave 2.1
grams of cream coloured putative 6-nitrobenzoxazolone. When on the hotplate, it starts to blacken at around 200C, melting at
240C. With the hotplate at 250C a sample melts instantly (literature 240C). Both melting point and yield of the 6-nitrobenzoxazolone is much
higher using the nitric/sulfuric acid nitration instead of using 65% nitric at 50C, suggesting that either less oxidation is occuring this way, or
this nitration scheme is more selective towards 6-nitro instead of the much lower melting 5-nitro isomer.
Reduction of 6-benzoxazolone to 6-amino benzoxazolone:
The reduction of 6-nitro benzoxazolone was performed as described in some article I found. To 70 ml water was added 2 ml of 8% HCl and brought to 90C.
Then 6 grams of steelwool was added in slightly compacted chunks. Over the course of 30 minutes, 2.1 grams of 6-nitrobenzoxazolone was added in small
portions. The dissapearance of yellow colour is a good indication that complete reduction has occured, which only took about 45 minutes. Then 2.5
grams of NaOH in 10 ml water were added, everything heated to 50 deg and the iron oxides filtered off. The basified filtrate containing the sodium
salt of 6-aminobenzoxazolone turned slighly reddish over the course of several hours. The filtrate was neutralized with dilute HCl to precipitate the
6-amino benzoxaolone. This needs to be done carfully with good lighting to precipitate most of the product. If the pH is lowered little too much,
everything goes immediately in solution again. Final yield was 1.2 grams of brownish/white glittering crystals that melted sharply at around 200C
(literature 202C), brownish colour probably from sligth oxidation or iron contamination. Product was not recrystallized.
|
I just want to point out that into the document you posted a few post above "Synthesis of some substitued benzoxazolone" they mention on p2:
QUOTE:
6-Nitrobenzoxazolone was prepared using the method of St. Von Chelmi~ki.~ Fifty grams of benzoxazolone was added in portions to 240 ml. of concd.
nitric acid. Slight warming was necessary to start the reaction, then it was kept at 50" by addition of benzoxazolone. Product began to separate
toward the end of the addition. After standing 20 minutes the mixture was diluted with water to give 55 g. of 6-nitrobenzoxazolone. Recrystallization
from ethanol-water raised the m.p. to 146'.
6-Aminobenzoxazolone.-The above nitro compound was hydrogenated in 70% alcohol using 570 palladium-on-Darco. It was crystallized from methanol, m.p. 310". Anal. Calcd. for C7H&*O*.HCl: C, 45.05; H, 3.78. Found: C, 45.25; H, 4.00.
END OF QUOTE:
So between your 240°C and the reported 146°C there is a very big gap...one of the two must be wrong somewhere.
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks for that drawing, it does make things more clearly. There is indeed
something funny about the melting points mentioned in the various nitration schemes of benzoxazolone in literature. Maybe using high temperature 65%
nitric, partial hydrolysis happens during the reaction, producing more of the 5-isomer, leading to melting point depression, or maybe has to do with
the reasonance of the oxazolone group and acidity during reaction otherwise. I do know that the melting point of 6-nitrobenzoxazolone from commercial
providers is given as 244-249 C, and 6-aminobenzoxazolone as 202 C, so these are more trustworthy probably.
I don't think there was much left after the nitration of 6-amino benzoxazolone, even very small amounts of some residual nitrophenols could produce
the orange colour and if there would have been anything left, even it's possible ammonium, sodium and potassium salts must have also been extremely
soluble. I lack any good extraction solvents like ether or DCM, so wouldn't have been an option anyway.
Your right, o-aminophenols (and m-aminophenols) are quite easily destroyed (even in the presence of deactivating groups), so probably nitration of
2-aminophenols even when partly nitrated wont work. Quite surprising that this is very different from p-aminophenols indeed. In fact, p-aminophenol
itself can actually be nitrated using mixed acids in reasonable yields, in which case a sulfonic acid and nitro group are added ortho to the hydroxy
group [1]. In contrast to acetaminophenol, benzoxazolone does not seem to react with N2O3 or N2O4 at ordinary condidions to produce an N,N
acetylnitroso and rearrangments to the diazoacetate, leading to deactylation/deprotection. [2]. So at least for benzoxazolone itself, the
o-aminophenol groups seem well protected during nitration. The question is what happens when an amino group is added, which can be oxidized and may be
stabilized by the lactim form. I was hoping that using 65% nitric this would favor formation of a 6-quinone. The position and order of the nitrogroups
introduced would also matter and I'm not into organic chemistry enough to make any acurate predictions. A nitro in 7 position would likely lead to
ring opening [3], and the possible resulting formamido group may react with HNO2, maybe leading to deamination with evolotion of N2 and CO2, or other
side reactions. For that matter nitration of 6-chloro benzoxazolone might be interesting, perhaps leading to a diazooxide or 3-chloro 2,4,6
trinitrophenol directly. Then again, maybe I'm missing something and the nitration would stop at the 5-nitro 6-chloro benzoxazolone or would be
destroyed completely again. Chlorination of benzoxazolone itself should be pretty straightforward, with the most prominent danger being
over-chlorination. I noticed some russian patent that got 80-90% yields using bleach and HCl directly, though sulfuric acid/TCCA may also be an
option. I'm not overly fond of chlorinating and nitrating chlorinated benzoxazolones though and not sure I will actually do these experiments, due to
toxicity reasons. Maybe I tried nitrating the wrong amino-benzoxazolone isomer and maybe the 5-amino benzoxazolone would behave different during
nitration.
To come back to the DDNR and isoforms, there may be a good reason why TNR was reduced by stannous chloride instead of sulfides in the patent by von
Herz. Generally solvent and reducant used can affect the position of the nitro being reduced. Sulfides may be more selective for reduction of the
nitrogroup in between the OH groups, which would likely render it very sensitive to oxidation via formation of a pyrogallol intermediate. The only
other isoform I've seen described for DDNR is one having a 1,2 quinone structure, it synthesis should be listed in this article [4] though seems to be
missing from the available pages.
The ammonium molybdate mediated nitration of acetaminophenol would be another potential route [5]. The only way I could see this working is if the
molybdate forms some very stable complex with the hydroxy group of acetaminophen, enough to prevent any oxidation. Though even in this case the
authors would have found di and trininitro derivatives or difficulties in the workup, so I think it is bogus. Another thing is that I found multiple
occasions were 2-nitro-4-acetminophenol was wrongly denoted as the 3-nitro 4-acetminophenol. I wouldn't mind someone proving me wrong though.
[1] Riegel, E. Raymond, Howard W. Post, and E. Emmet Reid. "THE NITRATION OF SUBSTITUTED ANILINES1." Journal of the American Chemical Society 51.2
(1929): 505-508.
[2] Baker, John K. "Nitrosation of benzoxazolinone." Bulletin of environmental contamination and toxicology 16.6 (1976): 743-745.
[3] Zinner, Helmut, et al. "Benzazole, IX. Über das chemische Verhalten der Nitro‐benzoxazolone." Chemische Berichte 92.2 (1959): 407-414.
[4] Heller, Gustav, and Theodor Hemmer. "Chinonbildung aus Nitroacetaminohydrochinon." Journal für Praktische Chemie 129.1 (1931): 207-210.
[5] Sana, Sariah, et al. "Mild, Efficient and Selective Nitration of Anilides, Non-Activated and Moderately Activated Aromatic Compounds with Ammonium
Molybdate and Nitric Acid as a New Nitrating Agent." Chemistry Letters 1 (2000): 48-49.
[Edited on 19-11-2016 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Would still like to try the nitration of 2 nitro-4-acetaminophenol using 65% nitric, though I'm out of nitric. A while back I came across an article
describing the nitration of 2-carboxy 4-acetaminophenol using 25% nitric (attachment). The author assumes the compound is a 2-nitro, though the
exceedingly high melting point may suggest a 3-nitro (assuming no oxidation producs have formed).
No luck with the nitration of 6-chloro benzoxazolone, Im adding it here just for the sake of it:
6-chloro benzoxazolone:
1 gram of finely powdered benzoxazolone was added to a 50 ml erlenmeyer, containing 30 ml's of water and heated to 70 deg C. Then, 6 grams of 8% HCl
was added. Stirring was continued for 5 minutes, upon which the benzoxazolone partly dissolved. The flask was lightly stoppered and over the course
of about 30 minutes, 0.55 grams of finely powdered TCCA was added in small portions, directly stoppering the flask again afterwards. Good stiring is
required (vortex) or the TCCA will only remain on top of the solution. The liquid turns greenish directly after the addditions, which fades to
almost colourless after only 5 minutes or so. A fine precipitate will become visible after each addition (cyanuric acid and the more insoluble
chloro-benzoxazolones (This may prevent large amounts of overchlorinated products ) Stirring was continued for another 30 minutes and then allowed to
cool to room temperature. The contents were filtered and washed several times,. The semi-dry solid was directly transfered to a 50 ml beaker, and 20
ml of 95% ethanol were added. While keeping at around 70 deg C, water was added untill slight clouding could be observed (6-7 ml). The solution was
then heated to boiling and slowly allowed to cool to room temperature. The colourless , shard like, crystals washed and collected. The melting point
is estimated between 185-195 (hotplate), with some sublimation visible. Doesn't melt sharply, probably also an unknown amount of 5-chloro present.
Total yield was 0.7 grams. (With stronger cooling more could have been precipiated, though I didn't want any cyanuric acid to also precipiate)
Nitration of 6-chloro benzoxazolone:
0.5 grams of 6-chlorobenzoxazolone was added to 10 ml sulfuric acid and cooled on ice. Over the course of 15 minutes, 0.71 grams of AN (dissolved in 2
grams SA) was added. Immediately, the colour changed to a beautiful golden orange. After 30 minutes or so, an off-white product started to
precipitate. Raising temperature above 40 reulsted in very slight gas production and only slighly more dissolution. Melting point of precipitated
product around 250C, probably 6-chloro-5-nitro benzoxazolone (couldn't find Tm in literature).

One other idea I had was to acetylate the 6-aminobenzoxazolone using heating with acetamide in closed system (possible?)
[Edited on 29-11-2016 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by nitro-genes | Would still like to try the nitration of 2 nitro-4-acetaminophenol using 65% nitric, though I'm out of nitric. A while back I came across an article
describing the nitration of 2-carboxy 4-acetaminophenol using 25% nitric (attachment). The author assumes the compound is a 2-nitro, though the
exceedingly high melting point may suggest a 3-nitro (assuming no oxidation producs have formed).
No luck with the nitration of 6-chloro benzoxazolone, Im adding it here just for the sake of it:
6-chloro benzoxazolone:
1 gram of finely powdered benzoxazolone was added to a 50 ml erlenmeyer, containing 30 ml's of water and heated to 70 deg C. Then, 6 grams of 8% HCl
was added. Stirring was continued for 5 minutes, upon which the benzoxazolone partly dissolved. The flask was lightly stoppered and over the course
of about 30 minutes, 0.55 grams of finely powdered TCCA was added in small portions, directly stoppering the flask again afterwards. Good stiring is
required (vortex) or the TCCA will only remain on top of the solution. The liquid turns greenish directly after the addditions, which fades to
almost colourless after only 5 minutes or so. A fine precipitate will become visible after each addition (cyanuric acid and the more insoluble
chloro-benzoxazolones (This may prevent large amounts of overchlorinated products ) Stirring was continued for another 30 minutes and then allowed to
cool to room temperature. The contents were filtered and washed several times,. The semi-dry solid was directly transfered to a 50 ml beaker, and 20
ml of 95% ethanol were added. While keeping at around 70 deg C, water was added untill slight clouding could be observed (6-7 ml). The solution was
then heated to boiling and slowly allowed to cool to room temperature. The colourless , shard like, crystals washed and collected. The melting point
is estimated between 185-195 (hotplate), with some sublimation visible. Doesn't melt sharply, probably also an unknown amount of 5-chloro present.
Total yield was 0.7 grams. (With stronger cooling more could have been precipiated, though I didn't want any cyanuric acid to also precipiate)
Nitration of 6-chloro benzoxazolone:
0.5 grams of 6-chlorobenzoxazolone was added to 10 ml sulfuric acid and cooled on ice. Over the course of 15 minutes, 0.71 grams of AN (dissolved in 2
grams SA) was added. Immediately, the colour changed to a beautiful golden orange. After 30 minutes or so, an off-white product started to
precipitate. Raising temperature above 40 reulsted in very slight gas production and only slighly more dissolution. Melting point of precipitated
product around 250C, probably 6-chloro-5-nitro benzoxazolone (couldn't find Tm in literature).
One other idea I had was to acetylate the 6-aminobenzoxazolone using heating with acetamide in closed system (possible?)
[Edited on 29-11-2016 by nitro-genes] |
You wrote no luck with the nitration...but you got something...so it is a partial succes. The only trouble is knowing for sure what you got.
To acetylate the 6-aminobenzoxazolone with acetamide...it must not be done into a closed system but under reflux...to allow for the volatile NH3 to
escape and displace the equilibrium of the transamidation.
Ar-NH2 + H2N-CO-CH3 <--==> Ar-NH-CO-CH3 + NH3(g)
In principle heating the the 6-aminobenzoxazolone with acetic acid should make the dehydration of 6-aminobenzoxazolone acetate into the
6-acetaminobenzoxazolone.
CH3-CO-OH.H2N-Ar --heat--> CH3-CO-NH-Ar + H2O(g)
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Something all right... I don't trust my chemistry instincts enough to tell what most likely formed, so I didn't fiddle with it too much. From a small
burn test (outside) I would say probably 1 nitro was introduced, likely in 5 position. Then again, I'm not even sure how the oxazolone ring behaves
in SA of higher percentages... inactivation, ring opening, something else? Quit complex indeed, let alone which positions are activated with other
functional groups attached. Hard to tell exactly what products form only by observing the colour, taking a very rough melting point and holding a
lighter to it.
[Edited on 3-12-2016 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
@Nitro-genes,
Just a quick question.
How are you sure about the 6- into your 6-chlorobenzoxazolone?
Came your procedure from a validated synthesis?
Otherwise you could have 4,5,6 or 7 halogenation... or even multiple halogenation (what would lead to a messy mix and unclear (not sharp) melting
point).
The only way to be sure of the position would be to perform an ipso replacement of the NH2 by diazotation via Sandmeyer's (the NH2 would then have to
be done from reduction of known NO2 compound (6-nitrobenzoxazolone) via SnCl2/HCl or Fe/HCl).
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Possible that multiple isomers were formed, although the preference for the 5 and 6 positions are almost universal. This was sort of an untested,
unsubscribed procedure indeed, something I should have mentioned. It was loosely based on 2 procedures: one describing chlorination of benzoxazolone
using hypochlorite and chlorinated solvent at Ph 1-2, and patent US 4435577, describing chlorination of benzoxazolone using chlorine gas at 40-80 deg
C and dioxane as solvent. I reckoned that hydrolysis at this temperature and acid concentration would not be a big issue. I was curious whether
without solvent chlorination would still work, reckoning that precipitation of the chlorobenzoxazolone could prevent over chlorination. Perhaps
running at a lower temperature would be better. I must say, the crystals that formed looked very good, having a very similar appearance to
recrystallized picric acid from a saturated water solution, only completely colourless. Are there any examples were multiple isomers can
co-crystallize like that? I always sort of assumed that if well defined crystals formed, it would be most likely an isomeric ally pure compound.
[Edited on 3-12-2016 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by nitro-genes | Possible that multiple isomers were formed, although the preference for the 5 and 6 positions are almost universal. This was sort of an untested,
unsubscribed procedure indeed, something I should have mentioned. It was loosely based on 2 procedures: one describing chlorination of benzoxazolone
using hypochlorite and chlorinated solvent at Ph 1-2, and patent US 4435577, describing chlorination of benzoxazolone using chlorine gas at 40-80 deg
C and dioxane as solvent. I reckoned that hydrolysis at this temperature and acid concentration would not be a big issue. I was curious whether
without solvent chlorination would still work, reckoning that precipitation of the chlorobenzoxazolone could prevent over chlorination. Perhaps
running at a lower temperature would be better. I must say, the crystals that formed looked very good, having a very similar appearance to
recrystallized picric acid from a saturated water solution, only completely colourless. Are there any examples were multiple isomers can
co-crystallize like that? I always sort of assumed that if well defined crystals formed, it would be most likely an isomeric ally pure compound.
[Edited on 3-12-2016 by nitro-genes] |
OK nice hypothesis of work.
To be sure of 1 isomer.
Take native sample --> a minute amount sample S0
Recrystalize the rest --> a minute amount sample S1
Recrystalize the rest --> a minute amount sample S2
Make a melting test of all 3 samples in close viccinity.
S0 should melt very close to S1 and S2 if there is a big gap of superior or equal to 10°C between S0 and
S2; then sample is unpure. Normaly 0 should meld prior to 1 itself prior to 2.
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
When I was reading Merck Index to find interesting molecular structure; I fall onto this informations:
5-chlorobenzoxazolone
CAS: 95-25-0
Formula: C7H4ClNO2
Molecular weight: 169,565
Use as a drug:
Muscular relaxant under the names:
Biomioran
Chloroxazone
Chlorzoxazon
Escoflex
Mioran
Miotran
Myoflexin
Myoflexine
Neoflex
Paraflex
Pathorysin
Solaxin
5-chloro-2(3H)-Benzoxazolone
5-chloro-2-Benzoxazolinone
2-Hydroxy-5-chlorobenzoxazole
5-Chloro-2-benzoxazolinone
5-Chloro-2-benzoxazolol
5-Chloro-2-benzoxazolone
5-Chloro-2-hydroxybenzoxazole
5-Chlorobenzoxazolidone
5-Chlorobenzoxazolinone
USAF MA-10
5-Chlorbenzoxazolin-2-on
5-Chloro-3(H)-2-benzoxazolone
5-Chlorobenzoxazol-2-one
5-Chlorobenzoksazolon-2
5-Chlorobenzoksazolinon-2
Flexazone
Parafon Forte DSC
NSC 26189
Solubilities:
Weakly soluble into water;
Soluble into methanol, ethanol and isopropanol;
Soluble into NaOH or NH4OH solutions
Melting point:
191-191,5°C
Synthesis:
See US Patent 2.895.877
Chemical structure:
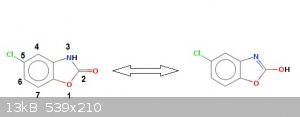
Here follows the patent.
Attachment: US Patent 2895877 synthesis 5-chlorobenzoxazolone.pdf (721kB)
This file has been downloaded 505 times
It discloses amongst others:
1°) the synthesis of 5-chloro compound ...
but from hydrolysis of a 5-chloro-2-aminobenzoxazole (the -N=C(OH)-O- sequence comes from a -N=C(-NH2)-O- one) --> Pretty useless for us.
or from 4-chloro-2-amino-phenol and phosgene (p-chloro-2-amino-phenol) --> nothing new under the sun but more OTC-ish if instead of toxic phosgene,
urea or dimethylcarbonate ester are used.
2°) the synthesis of the 6-chloro compound from benzoxazolone and Cl2 with a mp of 196°C.
It is also a myo-relaxant.
[Edited on 13-12-2016 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks Philou, it seems the melting point is in range of what I found for the chlorinated benzoxazolone. Read something about it's use as muscle
relaxant, though I'm not inclined to eat anything I produce in my shed.
Ok, here is something strange... This is based on 1 rushed experiment, it would
need further examination, but I'll add it here since I don't store these things on my computer.
I was bored and had a lot of purified isopicramic acid left and decided to look at what exactly forms during nitration using 97% SA and a nitrate
salt. To a 20 ml beaker, 10 grams of 97% SA was added and 0.5 grams purified isopicramic acid added and stirred at 20 deg C until everything
dissolved, producing an almost red/black but transparent solution. Next, it was added to an icebath and at 0 deg C, 0.25 grams of KNO3 (~1 mol eqvt)
was added. The solution was allowed to stir and no gas evolution was seen. Gradually, the solution took on a much lighter red transparent colour,
indicating something was happening. A small sample withdrawn at this stage dissolved in icecold water completely, not a single bubble of gas was
produced. Since this could be exlained by the H2SO4 adduct with isopicramic itself, I decided to add another mole equivalent of KNO3 and very soon,
gas formation became evident. It was allowed to stir overnight in the icebath, going from 0-10 degrees, with steady evolution of gas. When water was
added in the morning, copious amounts of NOx were liberated and a transpararent yellow/orange solution was left. No precipitate occured when kept at 4
deg C overnight. About 3 ml's of ethyl acetate were added and briefly stirred, upon which almost all of the colour transfered to the organic phase.
This was siphoned off and allowed to evaporate. A small amount of a yellow/orange crystalline precipitate formed, that burned very characteristic for
a diazonium compound, very vigorous (more than p-DDNP) and with yellow flash. It dissolved very easily in water again, but adding a saturated KNO3
solution and chilling produced no precipitate. It further seems to attack metals like crazy, although this could also be due to some extracted acid by
the ethylacetate.
So, what formed here? Since it is water soluble, even after extraction, it is
not p-DDNP iself, since the diazonium sulfate salt dissociates very quickly upon dilution. It still contains a diazonium group, but since it does not
produce a precipitate with KNO3, it likely also isn;t DDNR, since the K-salt is reported to be very insoluble (DDNR istelf as well). One of the
options is that a 1,2 quinone 3,6 dinitro 4-diazo is formed due to hydrolysis of the 2-nitro of isopicramic acid, though I'm not sure this would
explain the reactivity towards metals. Another option is the 2,3,6 trinitro 4-diazo phenol descirbed before, though this would be strange considering
the described deactivation of the amine group in 97% SA. Other IMO, less likely options would be some sulfonic acid replacement of the nitro, or more
likely maybe some azoxy compound from coupling reactions. Any guesses, anyone?
[Edited on 17-12-2016 by nitro-genes]
|
|
|
| Pages:
1
..
22
23
24
25
26
..
33 |
|








