nlegaux
Hazard to Self

Posts: 93
Registered: 28-11-2014
Location: East Tennessee
Member Is Offline
Mood: No Mood
|
|
Preparation of 2-aminophenol from Salicylic Acid
Presented below is a useful procedure for the production of salicylamide, benzoxazolone, and ultimately 2-aminophenol for the home chemist. It
utilizes mostly OTC materials in order to accomplish this.
A. Materials
-Salicylic Acid
-Carbamide
-Boric Acid
-10M Hydrochloric Acid
-1M Hydrochloric Acid
-Household Bleach
-10% aqueous ammonia
-3g sodium hydroxide
-10% aqueous sodium hydroxide
B. Procedure
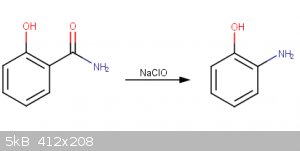
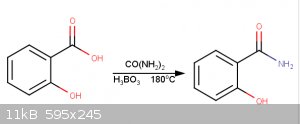
This procedure begins with the synthesis of salicylamide from salicylic acid (1).
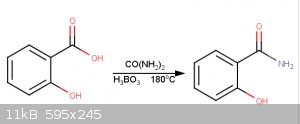
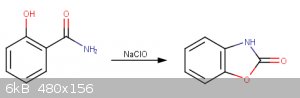
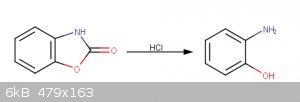
Following this, a Hofmann rearrangement reaction is performed to create 4,5-benzoxazolone, which is hydrolyzed with dilute HCl(aq) to 2-aminophenol.
Preparation of Salicylamide:
10.5g salicylic acid is combined with 13.6g of carbamide and 0.8g of boric acid, to act as a catalyst, in a 125mL Erlenmeyer flask. The Erlenmeyer
flask is connected to a water trap. The melt is heated to 180°C for 2 hours using a sand bath. 50ml water and 5ml 10% aqueous ammonia added. Solution
boiled for 5 minutes. Solution chilled and 25ml 10M HCl(aq) added. Salicylamide filtered off.
The final product was a white crystalline powder with mass of 3.2g, representing a 31% yield. On future trials, 40mL of 10M HCl(aq) was used,
increasing the yield to 89%. The melting point of the powder was found to be 141°C (lit. 140-144°C) using the melting point apparatus from source 5.
This product forms a purple complex with FeCl3, but it is not nearly as intense as the complex formed with Salicylic acid. This indicates
that the product is contaminated with Salicylic acid, which is to be expected.
Preparation of 2-aminophenol
1.7g salicylamide added to 50mL H20 in a 125mL beaker. A solution comprised of 13mL household bleach, 3g NaOH, and 20ml H2O is
also prepared. The NaClO solution is added added to the salicylamide solution drop wise in an ice bath with stirring. After complete addition of the
NaClO, 15ml 10% NaOH is added and the temperature of the solution is rapidly increased to 70°C for 30 minutes. The solution is next cooled with an
ice bath, and 25mL 10M HCl(aq) is added. This causes the benzoxazolone intermediate to precipitate, which is dried at 100°C for one hour. This
product is next dissolved in 100ml 1M HCl(aq) and heated to 80°C with reflux for 1 hour. The solution is allowed to cool to room temperature and the
product is filtered.
The product first isolated in the procedure, most likely benzoxazolone, has a melting point of 141°C (lit. 139-142°C) and a mass of 1g. The second
product of 2-aminophenol has a mass of 0.25g and a melting point of 173°C (lit. 174°C).
C. Discussion
The 2-aminophenol from this procedure can be used in the manufacture of (poly)2-aminophenol (2) and a broad range of heterocylic compounds, including
benzoxazoles (3).
D. Sources
(1) Smuv. http://www.sciencemadness.org/talk/viewthread.php?tid=4201&a...
(2) B. L. Rivas, et al. Polymer Bulletin. December 2002, Volume 49, Issue 4, pp 257-264. "Synthesis, characterization, and properties of poly(2- and
3-aminophenol) and poly(2- and 3-aminophenol)-Cu(II) materials".
(3) Mitchell, S.C. & Waring, R.H. "Aminophenols." In Ullmann’s Encyclopedia of Industrial Chemistry
(4) Bachmann, Werner E. et al. Organic Reactions Volume III. p.277. http://library.sciencemadness.org/library/books/organic_reac...
(5) bob800. http://www.sciencemadness.org/talk/viewthread.php?tid=25138
[Edited on 9-13-2015 by nlegaux]
[Edited on 9-13-2015 by nlegaux]
|
|
|
AvBaeyer
National Hazard
Posts: 644
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
1. How pure was the salicylamide that you made? How was it characterized if at all?
2. Why such a broad melting range for your o-aminophenol? Why is it considerably different from the reported value? Are you sure that you ran the
Hofmann rearrangement properly?
3. For example, did you consider that there may have also been ring chlorination during the Hofmann reaction?
Overall, you need to provide convincing data that your synthesis worked as desired at each step.
AvB
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
I agree with AvBaeyer that more characterization of the products would be nice. A visual assessment of the color and crystalline structure of the
product is useful, alternatively add a picture. I've run the boric acid catalyzed amide formation many times on a variety of substrates, and every
single time a significant amount of polymerized crap is formed from the hot urea. Boiling with dilute ammonia water, vacuum filtration while hot if
necessary, then slow cooling and crystallization yields a reasonably pure product and is necessary before proceeding to the next step in a synthesis.
Also, you didn't use any alkali for the Hofmann rearrangement? The reaction requires two equivalents of alkali to proceed smoothly. Finally, did you
titrate the sodium hypochlorite or just use 8.25% bleach as is? If the latter, you should know that 8.25% doesn't correspond to 1.5M NaOCl, it can
vary wildly but is typically much closer to 1M NaOCl.
|
|
|
nlegaux
Hazard to Self
Posts: 93
Registered: 28-11-2014
Location: East Tennessee
Member Is Offline
Mood: No Mood
|
|
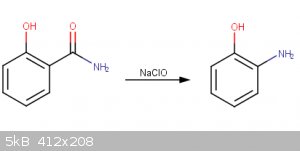
Thank you for these pointers, I will work on improving/correcting the report. The final product is at first a white crystalline solid, but browns over
time. When recrystallized, it creates a deep red solution, similar to the one seen in the second image, both of which which correlate with the data I
have found.
Commercial bleach is stabilized with sodium hydroxide, and I was under the impression that this would be sufficient to push the reaction to the
correct side (https://www.thecloroxcompany.com/products/sds/). I will rerun the synthesis with adjusted amounts of hypochlorite and with the addition of extra
NaOH, now that you have pointed out to me that 8.35% bleach is not 1.5M.
I will begin collecting the tools necessary to do a more accurate melting point analysis. The reason for the large range on the current melting point
presented is the incredibly crude melting point set apparatus I used (a sand bath with a watch glass sitting on top of it containing the sample). I
found that the temperature gradient across this setup is 10C, so I factored that into the final number.
When I have time in the future (probably this weekend) I will further characterize the product and include this characterization at the end of the
report, along with some additional photos.
nlegaux
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by nlegaux  | | The final product is a brown crystalline mass. Its melting point has been determined to be 180±10C, which correlates to 2-aminophenol's melting point
of 174C. An 86% yield was obtained. The compound readily sublimates forming white needle-like crystals. |
That is equivalent to nearly zero characterization.
Firstly, you need to report the melting point range. A ±10 °C is not a proper way to do that: What does that even mean? At which point does the
melting begin and at which point everything is melted? This requires two definite temperatures. What are the experimental conditions of the
measurement? The only information your value provides us is that your product is most likely not 2-aminophenol, as the mp should be equal or lower
than 174 °C (mp depression). Due to the nature of phase transitions in mixtures, the mp is only exceptionally higher than the lower mp component (it
requires the higher mp component to not be soluble in the melt of the lower mp component, which is rarely the case for closely related products).
Secondly, a melting point measurement, unless it has a very narrow range, is not enough in this specific case. The reason is in that all the more
probable products have melting points near 180 °C (e.g., biuret, 3- and 5-chlorosalicylic acid, 3- and 5-chlorosalicylamide). What you need is a
proper characterization of the purity and identity of the product. Purity can be indirectly estimated from the mp range, but more properly so by
chromatographic methods such as TLC (assuming you have no access to any spectroscopic instruments). Identity requires chemical characterization
besides the mp, because, as mentioned, other products have melting points in vicinity.
Thirdly, you present your synthesis as a one-pot amidation with urea followed by the Hofmann rearrangement. This obviously cannot work as such,
because besides the numerous reactive side-products, there is excess urea, urea self-condensation products and ammonia present. Any will happily react
with hypochlorite. Essentially, you present no evidence whatsoever that the amidation worked. You based your procedure on Smuv's report where the
amidation was done on benzoic acid and not salicylic acid. Did you at least check with TLC or anything at all? Or have you done an amidation on
salicylic acid separately?
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
nlegaux
Hazard to Self
Posts: 93
Registered: 28-11-2014
Location: East Tennessee
Member Is Offline
Mood: No Mood
|
|
Is this better characterization of the salicylamide intermediate?
"The final product was a white crystalline powder with mass of 3.2g, representing a 30.6% yield. The melting point of the powder was found to be 141C
(lit. 140-144C) (5)."
I would do a TLC, but I do not currently have the resources to do so. Is there any other kind of characterization information I should include before
going ahead with the Hofmann rearrangement? If not, I will correct the later half of the procedure and perform the reaction soon.
nlegaux
[Edited on 8-22-2015 by nlegaux]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by nlegaux | Is this better characterization of the salicylamide intermediate?
"The final product was a white crystalline powder with mass of 3.2g, representing a 30.6% yield. The melting point of the powder was found to be 141C
(lit. 140-144C) (5)."
I would do a TLC, but I do not currently have the resources to do so. Is there any other kind of characterization information I should include before
going ahead with the Hofmann rearrangement? If not, I will correct the later half of the procedure and perform the reaction soon.
nlegaux |
It is better, but it is not optimal. For example, it gives no indication of purity as there is no range given and no description on how the
measurement was done. It makes a difference if it was just some roughly made measurement with a thermometer in a test tube or if it was done using a
Thiele method or a microscope.
But why don't you do a chemical characterization as well? For example, a FeCl3 tests comparison between the starting material and the
product would cost you nothing, but would give valuable information. Or whichever other test (like solubility test for ethanol, water, aqueous
NaHCO3, NaOH, HCl...).
In regard to your writing skills, I can give you these following few simple advices which you can follow without much effort. I believe following
these advices will not only improve the quality of your reports, but also help you understand how science works.
Pose your report in a scientifically meaningful way. A scientific report must have an introduction where the problem is defined and described, the
current state of the art reviewed and analyzed, and a solution is hypothesized. After the introduction you discuss your experimental design, your
results, their meaning and what new questions arose. After the discussion you provide the experimental. At the end it is always nice to provide a
conclusion that should refer to the hypothesis (Was it confirmed or overthrown? Are there additional discoveries? What is the scientific contribution
of your results? ...). In long reports an abstract at the beginning is always a good idea, but in short one like the present one it is unnecessary.
Note that the problem to be solved and a hypothesis can be something as simple as:
A preparatively useful route to salicylamide from easily available materials would be beneficial to the home chemistry community. Employing the
urea based amidation method on salicylic acid was considered as a potentially efficient route to salicylamide.
A literature review with a few relevant references is due, but it is a simple hypothesis so there is no need to go into too much detail.
And the conclusion and open issues can be equally simple:
The results indicate salicylamide can be obtained in low to moderate yields by heating salicylic acid with urea. Purity, reproducibility, side
products and optimal yields were not determined.
Use some scientific format for the experimental part (if you don't know how this is done, see any scientific article and practice in imitation):
Describing the procedure in bullets is certainly improper. It makes it look lousy. Many people will consider the excessive use of bullets in
any text as a sign of sloppiness or lack of understanding. The reason is in that bullets are useful to emphasize and declare an arbitrary
classification of issues about which you are discussing. But they should not be used for continuous and coherent flow of information (such as an
experimental procedure is). The other reason is in that the excessive use of bullets stereotypically correlates with the cluelessness of the authors
(particularly students and their reports). As objective as the reader tries to be, such stereotypes will always awake and judgments will be made based
on them. Note how I used bullets here to separate two distinguishable issues related to form.
Use proper units, blanks in front of the units and proper chemical names or shorthands. For example: Don't use formulas for nonexistent
compounds (such as NH4OH), or ambiguous formulas when you mean something else (like "HCl" which is a gas at rt, unlike "hydrochloric acid"
or "HCl(aq)" which is what you had in mind). For the last 35 years, the preferred symbol for liter is "L" (upper case) and not "l". You always put a
space between the value and unit. Thus, properly written it should be: "10.5 g salicylic acid", "180 °C", "5 mL 10% NH3(aq)", "25 mL 10M
HCl(aq)" (note that M is not a unit, but a concentration mark - when using a unit you use a blank as in "10 mol/L")... Also, use only meaningful
numbers and digits. For example a "30.6% yield" makes no sense. That should be rounded to 31%.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Interesting synthesis, thanks for posting!
I have a couple of questions, you write:
4- 50ml water and 5ml 10% NH4OH added
5- Solution boiled for 5 minutes
What exactly is adding the ammonia for, is it for solubilizing the cyanuric acid by products along with the benzamide formed? Have you meusured the pH
of the solution after the reaction and adding the water? I can imagine there is quite some ammonium carbonate there already from urea decomposition?
What is the boiling step for, wouldn't it lead to partial hydrolysis of the product?
Since I read about phosphoric acid as another catalyst, I was wondering how diammoniumphosphate (fertilizer, yeast nutrient, very cheap) would do
instead. It may have several benefits:
1. The added ammonia value may lead to less formation of urea self condensation products, such as cyanuric acid
2. Unlike phosphoric acid, diammonium phosphate is an anhydrous salt
3. 20% diammonium phosphate and 80% urea form an eutectic, melting at 120 deg C or so
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
| Quote: Originally posted by nlegaux |
4- 50ml water and 5ml 10% NH4OH added
5- Solution boiled for 5 minutes
6- Solution chilled and 25ml 10M HCl added
7- Salicylamide filtered off
|
I just noticed this. Why did you reacidify the solution before you filter it? One of the main purposes of boiling with an ammonia solution(you also
want a more concentrated solution of ammonia, about 1:5 if you solution is 10% although this depends on the pH of the solution, you want it to be
basic), is to remove unreacted acids. Boric acid, cyanuric acid, and salicylic acid all have a low solubility in acidic solutions, and thus you're
product may be contaminated with these acids. Acidic solutions can also protonate amides and increase their solubility, lowering the recovery of your
desired product.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
contemplating to perform this reaction, bit worried about toxicity of by-products during the synthesis at the various stages:
From literature, it seems probable that decarboxylation of salicylic acid also occurs at the temperatures utilized, since phenol boils at near the
reaction temperature, heating too hard will probably result in signifcant phenol formation escaping the reaction mixture by condensing or escaping,
maybe driving further formation of phenol? By the looks of it phenol can react directly with urea, though it requires a zinc catalyst and higher
temperatures. much less by products may form when the reaction is performed using ammonia salts and a high boiling polair aprotic solvent (which
unfortunately are not readility available OTC, with the possible exception of DMSO) 
The hoffman rearrangment using hypochlorite, even when performed with excess base, may still produce ring chlorinated by products, especially during
the last HCl acidification step. Some real monsters can be formed from chlorinated phenols:
https://en.wikipedia.org/wiki/2,3,7,8-Tetrachlorodibenzodiox...
Last but not least, is it possible phosgene gas can be formed from the reaction of benzoxazolone with hypochlorite?
[Edited on 28-8-2015 by nitro-genes]
|
|
|
nlegaux
Hazard to Self
Posts: 93
Registered: 28-11-2014
Location: East Tennessee
Member Is Offline
Mood: No Mood
|
|
The Diammonium phosphate idea isn't a bad one... Once I perfect the second stage of this synthesis, I may revisit the salicylamide synthesis step. I
did consider the formation of phosgene, but after reading up on it I read that it decomposes in water (http://www.inchem.org/documents/icsc/icsc/eics0007.htm), so I wasn't very concerned. As for the other byproducts, sufficient safety equipment
(goggles, gloves, etc...) should be enough protection.
The reason I reacidified the solution was to recover the Salicylamide. I am unsure of another way to collect the product (I'm open to ideas).
nlegaux
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Yeah, your right, any possible phosgene would immediately hydrolyse when the solution is kept well basified during the reaction with hypochlorite.
stupid, I'm a bit chlorophobic though On the other hand, phosgene is almost
insoluble in water and the hydrolysis of benzoxazolone using HCl could result in some phosgene formation, as it's hydrolysis in water is comparetively
slow. Maybe analogous to the synthesis described below:
http://www.molbase.com/en/synthesis_75-44-5-moldata-27616_35...
"selected from the synthesis mentioned above:
Phosgene. In this novel preparation, hydrogen chloride (4.2 g,115 mmol) was condensed onto N ,N 9-carbonyldiimidazole (2.5g, 15 mmol) at -196 C.
The reaction vessel was warmed to-63 C, and connected to a trap held at -196 C. The vesselwas opened and the excess of hydrogen chloride condensed
intothe -196 C trap. The gas-phase infrared spectrum of the con-tents of the -63 C trap showed strong bands at 1827 and 849cm 21 due to phosgene, as
well as a much weaker band at 2886cm 21 due to hydrogen chloride. Yield (based on N ,N 9-carbonyldiimidazole) 0.74 g (49.8%)."
I'll leave it to the experts in organic chemistry, but it may be safer to perform the hydrolysis of benzoxazolone using dilute sulfuric acid,
I wouldn't want to do this reaction with concentrated HCl! 
Regarding the hoffman degradationn, are you using one related to the one described here:
Process for the preparation of benzoxazolone, Patent CA 1150276 A1
http://www.google.com/patents/CA1150276A1?cl=en
Yields mentioned are up to 96% and the reaction can be performed at ambient temperature. It mentions that when kept well basified and the hypochlorite
is added slowly, ring chlorination is almost absent. Difficulty is probably determining the hypochlorite concentration, as the patent roughly uses
equimolar quantitites of hypochlorite and salicylamide. Too little, salicylamide will be present (same MP and roughly same solubilities), use of
excess unknown.
In the patent, any presence of residual hypochlorite is destroyed before HCl is added, which is presumably important to prevent ring chlorinated by
products. Ascorbic acid may be an even more OTC option for this. Looks doable, though purification and identification looks difficult.
For the purification of salicylamide from the melt, no really good ideas. Maybe phenol by-products can be removed by washing with or recrystallizing
from cold 10-20% ethanol or something. Maybe you can exploit somehow the difference in acidity between salicylic acid, cyanuric and the salicylamide,
like you did. Cyanuric acid, is almost completely insoluble in acetone, water and cold ethanol, so that may also be an option. Maybe ethanol/water
mixtures may be any good?
[Edited on 29-8-2015 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Great find for this synthesis route, it works in high yield, even with household bleach. The plan was to add the bleach slowly to the basified salicylamide solution, but I kind of panicked when I smelled something that
resembled green corn, just added all of the remaining bleach at once and left the reaction. Chlorine can produce a lot of nasty smells though so I'm
probably panicking, but did you smell the same thing?
 
|
|
|
nlegaux
Hazard to Self
Posts: 93
Registered: 28-11-2014
Location: East Tennessee
Member Is Offline
Mood: No Mood
|
|
Nice find on the patent nitro-genes! It offered a lot of insight. I have been isolating the benzoxazolone intermediate instead of the 2-aminophenol
product with the procedure I am currently attempting. In the near future I will attempt the hydrolysis with a stronger acid. Probably sulfuric acid,
as you suggested.
The first few times I attempted the reaction without cooling I also noticed an unusual smell. I have found that this smell is not present when an ice
bath is utilized.
nlegaux
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Yes, really surprised to find that the reaction described in the patent seems to work exactly as described. Like the patent suggested, KOH was used
for dissolving the salicylamide, have you tried the reaction with only NaOH? Was wondering if the potassium salt of salicylamide was really that much
more soluble than the sodium salt. Surpised also how pure the benzoxazolone seems directly from the hoffman degradation. For the recrystallization I
used 150 ml of water and 50 ml 96% ethanol @ 70 deg C, --> -10 it seems very effective in removing (chloro)phenol-like smells.
Regarding the smell, yes, it is a very peculiar one, noticeably different from either phenol or chlorophenol, both of which smell rather medicinal
(especially chlorophenol) or the biting smell of a chloramine. It was only present immediately after the hypochlorite additions and to me resembled a
fresh corn like smell. Good to know it is absent when performed in an ice bath.
How long do you reflux and with what acid srength btw? Curious how easiliy or difficult benzoxazolone hydrolyses.
[Edited on 2-9-2015 by nitro-genes]
|
|
|
nlegaux
Hazard to Self
Posts: 93
Registered: 28-11-2014
Location: East Tennessee
Member Is Offline
Mood: No Mood
|
|
I attempted the acid hydrolysis yesterday. I added 3ml 96% H2SO4 to 0.5g benzoxazolone and refluxed for 30 minutes. Unfortunately, I don't know the
effectiveness of this hydrolysis, because I broke my thermometer, and my new one doesn't come in the mail until the end of this week  . .
I read up on benzoxazolone hydrolysis a bit, and perhaps a base hydrolysis would be more suitable? This patent describes the hydrolysis of a
benzoxazolone derivative with NaOH. https://www.google.com/patents/US2979503
nlegaux
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
ortho and para aminophenols are rapidly oxidized under basic conditions leading to dark colored product and lots of tar.
|
|
|
nlegaux
Hazard to Self
Posts: 93
Registered: 28-11-2014
Location: East Tennessee
Member Is Offline
Mood: No Mood
|
|
I have found hydrolysis with dilute (1M) HCl(aq) for around an hour with reflux to be a successful way to hydrolyze the product.
nelgaux
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
I wonder if acetylsalicylic acid can withstand the intense heating with urea and boric acid without being deacetylated. If it could, then that would
simplify workup tremendously, as the acidic proton would be temporarily removed. The product would thus be insoluble in the ammonia solution, and
would allow for better separation from the various by products. An acid hydrolysis is a necessary step anyway, so the acetyl group shouldn't add any
more hindrance, or complicate things. I have a feeling that the acetyl group will be removed under the conditions of the initial reaction though.
Perhaps the salicylamide can be acetylated during the workup?
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Just to keep all the information together:
Polyphosphoric Acid as a Reagent in Organic Chemistry. IV. Conversion of Aromatic Acids and their Derivatives to Amines - salicylic acid +
polyphosphoric acid + hydroxylamine -> benzoxazol-2(3H)-one (lossen rearrangement);
Ueber die Hofmann'sche Reaction (Ueberführung der Amide in Amine) - salicylamide + NaOCl -> benzoxazol-2(3H)-one
Preparation of Benzoxazolones from Halogenosalicylhydroxamic Acids and Ethyl Chloroformate - bulletin_de_lacademie_polonaise_des_sciences_1971_t19_s15
- few methods for benzoxazol-2(3H)-one preparation mentioned, including heating of hydroxamic derrivative of salicylic acid with formamide.
Caronna, G.; Palazzo, S.,Gazz. Chim.Ital., (1960) 90,1100 - no electronic version of this one, salicylic acid + NaN3 + conc H2SO4 ->
benzoxazol-2(3H)-one (schmidt reaction) at 0-5 °C for 3-48h, mentions unsubstituted, 5-Me- and 5-nitro- 2-hydroxybenzoic acid.
Attachment: bulletin_de_lacademie_polonaise_des_sciences_1971_t19_s15.pdf (3.5MB)
This file has been downloaded 951 times
[Edited on 24-1-2016 by byko3y]
|
|
|
gluon47
Hazard to Self
Posts: 81
Registered: 20-9-2015
Location: oceania
Member Is Offline
Mood: fluorinated and dying
|
|
I've been following this synthesis and I just finished making the benzoxazolone intermediate.
It seems there are a few concerns about phosgene formation during the hydrolysis of benzoxazolone to 2-aminophenol using Hydrochloric acid.
How dangerous is this really? Are dangerous amounts of phosgene likely to be produced?
I was planning on performing the hydrolysis soon.
reality is an illusion 
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by gluon47 | I've been following this synthesis and I just finished making the benzoxazolone intermediate.
It seems there are a few concerns about phosgene formation during the hydrolysis of benzoxazolone to 2-aminophenol using Hydrochloric acid.
How dangerous is this really? Are dangerous amounts of phosgene likely to be produced?
I was planning on performing the hydrolysis soon. |
0% chance in aqueous solution.
|
|
|










