DrDevice
Hazard to Self

Posts: 74
Registered: 19-3-2012
Member Is Offline
Mood: Incompatible with carbon based lifeforms
|
|
Preparation of sodium salt of 4-hydroxydiazosulfonate
One simple and high yielding experiment I have performed recently is the synthesis of:
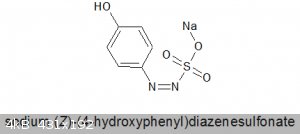
(MW = 224.17)
an intermediate on the way to hydrazinesulfonic acids.
Many texts refer to these as diazosulfonates, but ChemSketch (which I expect is more authorative then me  ) shows this as a diazenesulfonate. I'll keep calling them diazo compounds for convenience. ) shows this as a diazenesulfonate. I'll keep calling them diazo compounds for convenience.
A useful references is Saunders"The Aromatic Diazo Compounds". There are also useful texts by Zollinger, and a host of dye related books that also
touch on this, as the diazo are very important in early dye science.
PROCEDURE
Prepare a saturated solution of sodium sulfite with 18.42g (0.148 mol) in 40ml water, in a 600ml beaker. Not all the sodium sulfite will dissolve, and
without vigorous stirring during the addition, a hard lump will form. Some undissolved sulfite is OK, but it is important later in the reaction that
there are no large lumps. Heating the solution will help dissolve it all, but note that too much heating will degrade the sulfite releasing SO2. The
final result needs to be cooled and a slurry of crystals may re-form. This is OK, as there will be no large lumps. Refrigerate this solution.
Ammonium sulfite works equally well, and is much more soluble.
Prepare a solution of 5.155g (0.075 mol) sodium nitrite in 8ml water, and place in freezer. The exact molar ratio is critical here. More on this
later.
Place 8.00g (0.073 mol) of 4-aminophenol

in a 300ml wide mouth conical flask. I used a wide mouth flask because it is easier to then add the ~30g ice now required.
Add 19ml conc (12M) HCl = 0.228 mol, and stir in an ice-salt bath. A pink-grey slurry forms.
SLOWLY add drop by drop the sodium nitrite solution, with stirring. The solution will go a deep blue-purple colour immediately.
A burette is a convenient way of addition. Monitor the temperature of the solution to ensure it does not rise above 4C. You will find the ice starts
melting at a great rate as this reaction proceeds.
The sodium nitrite and hydrochloric acid forms nitrous acid in-situ. This reacts with the 4-aminophenol to form the diazonium:

According to the texts, excess nitrite apparently interferes severely with the next step. I can't say by how much, as I haven't experimented, but I
always stop adding nitrite solution before it is all consumed, and check the diazonium solution by adding a drop to a starch/iodide solution. Excess
nitrite will produce a deep purple colour instantly. Basically, towards the end, add a drop, then check. This can be tedious...Also as the reaction
comes to completion, reaction of the nitrite can be slow.
If you end up adding too much nitrite, it can be removed with sulfamic acid or urea. Sulfamic acid is much faster than urea.
When you have a compund that is "just right", it will produce a faint purple reaction in the starch/iodide.
By now, the walls of your flask will probably be spattered with a deep blue/purple solution. All the solids (4-AP, ice etc) should be
dissolved/liquid.
Take the sulfite solution from the fridge and start it stirring vigorously with a large magnetic stir bar. Check there are no large lumps of
undissolved sulfite.
Add the diazonium solution to the stirring sulfite solution all at once. A thick yellow precipitate should form within a few seconds. If ammonium
sulfite was used, it may thicken so much that it forms an unstirrable mass. Stir manually for a while and this should break up to allow the magnetic
stirrer to work.
Keep stirring for about 0.5hr. This gives any undissolved sulfite a chance to react.
Filter the result over suction, and press the resulting filter cake in the filter paper between a few layers of paper towel to remove as much liquid
& inorganic salts as possible.
The damp solid can be dried at 50C for a few hours, resulting in a fine, bright yellow powder that is very soluble in water and not soluble in
acetone, ethanol, methanol, ether or DCM.
From the 8.00g (0.0734) mol of 4-AP, I obtained 15.8g product, which is 96% yield...I'm not convinced that I actually got a yield this high - there
may be a fair amount of inorganic salts remaining from the filtering, but as the product is so water soluble, they are difficult to remove.
I do not have access to any analysis equipment that could confirm the structure, but the reaction & product colours are all as per textbook.
I have used a similar procedure successfully with varying yields with 4-methoxyaniline, and 3,5-dimethoxyaniline. I am trying to obtain
3,5-dihydroxyaniline (see my other thread) to perform similar experiments,
3-aminophenol and 3-methoxyaniline have not been succesful (black gloop  ) )
I'll post again soon with notes on reduction of this to hydrazine derivatives.
|
|
|
Paddywhacker
Hazard to Others
Posts: 478
Registered: 28-2-2009
Member Is Offline
Mood: No Mood
|
|
A nice writeup, thanks. What are your plans for the material?
|
|
|
DrDevice
Hazard to Self
Posts: 74
Registered: 19-3-2012
Member Is Offline
Mood: Incompatible with carbon based lifeforms
|
|
Thanks for the kind words, Paddywhacker.
Right now, I am at the "Oooh! Shiny!" stage of admiring the fruits of my labour and collecting various results of the reactions. 
But perhaps in the far far future, well, phenylhydrazines and Fischer indolization do tend to go hand in hand...but I have a way to go in skill
development yet.
|
|
|
|








