Archy12345
Harmless

Posts: 6
Registered: 4-3-2016
Member Is Offline
Mood: No Mood
|
|
Reduction of Methyl 3-Nitrobenzoate to Methyl 3-Amino Benzoate
So over the past week I have been trying to convert methyl 3-nitrobenzoate into methyl 3-aminobenzoate. I have not been very successful, though.
I am taking the iron powder/acetic acid route, and I read a paper not too long ago which described the reduction of nitro groups to amine with Fe/AcOH
using sonication instead of reflux. I will attach a link at the end of the post.
I am using a 5mL of a 2/2/1 Ethanol/Acetic Acid/Water mix, with 0.15g of my nitro compound and 0.23g of reduced iron powder (5 equivalents). I put all
this in a test tube and expose it to ultrasound for 1h. I occasionally stir the mixture vigorously. My ultra sound bath delivers 40kHz and anywhere
from 80W-130W of power. I'm not the owner of the machine, and I'm not entirely sure.
I can see that iron powder is being oxidized because of a brown powder forming in the test tube. So there is SOMETHING happening.
After the ultrasound exposure, I pour the contents of the test tube into a beaker and add about 20mL of aqueous 3M NaOH. This is for the purpose of
precipitating out all of the dissolved iron ions and increasing the pH to about 10 or 11. The addition of hydroxide creates something akin to a thin
slurry. It is notable to say that when made basic the solution acquired a grape-like smell. The target compound is a regioisomer of methyl
anthranilate, so perhaps this is a good sign. I pass this through a frit (which takes forever as you'd expect), then I wash the filtrand with 25mL of
ethyl acetate. The filtrate I get is clear and colorless and has two layers. This gets transferred into a separatory funnel and shaken/vented until
pressure no longer audible when venting. I then proceed to extract three more times with 25mL of ethyl acetate. The organic layers are combined and
dried with anhydrous sodium sulfate. The ethyl acetate is removed using a rotatory evaporator.
This step yields two out comes. One outcome is that there is nothing left once the ethyl acetate has been removed.
The second outcome is that there is a very viscous yellow liquid residue in the flask. This only happened once. I added some DCM to redissolve the
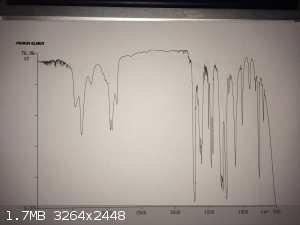
yellow liquid and transferred it into a vial. I ran an liquid film IR on my sample, which is attached. The link to an authentic IR is also provided at
the bottom of this post.
The IR isn't perfect, but it wasn't awful either. I set up an hydrogen chloride gas generator and gassed the contents of the vial. A very fine solid
precipitate did form. I centrifuged out the solid (the solid formed a pellet at the top of the solution, which I thought was interesting, but I
suppose it's not hard to be less dense than DCM). the DCM was pipetted into a beaker. I added more DCM to rinse the solid, and this was also
centrifuged. I pipetted as much of the DCM as I could, then transferred the contents of the vial into a round bottom flask and used the rotatory
evaporator to get rid of all the DCM. At this point I wish I had taken an IR of the solid by KBr pellet. I didn't, but I did weigh it, though. It's
mass was roughly 0.07g. Which corresponds to a terrible yield, assuming I had the right compound anyway.
I decided I wanted to try to convert the compound back into its freebase form. The solid readily dissolved in water, which is a good sign. I tried to
extract the salt via ethyl acetate, but upon evaporation the flask was empty.
So! Does anyone have any thoughts on any of this? I've run the reaction 5 times so far. I have obtained yield 2/5 times, and only one of those times
gave a decent IR. If anyone who has experience with this type of reaction, help is greatly appreciated. I thank you in advance.
Paper about Ultrasound:
http://ro.uow.edu.au/cgi/viewcontent.cgi?article=1174&co...
Authentic IR:
http://www.chemicalbook.com/Spectrum/4518-10-9_IR2.gif
|
|
|
Dr.Bob
International Hazard
Posts: 2658
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: No Mood
|
|
I seem to remember that some HCl helps this reaction. You could also try Zinc powder, granules or dust (even the dust from inside a large box of
galvanized nails might be suffucient), tin has also been used. All have their pros and cons. If you can get sodium dithionite, found in some OTC
rust removers, that also can reduce nitro groups. The simple way is Pd/C in ethanol and H2, but that is not very OTC, however, many variations
exist.
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
If you give up on saving the ester, 3-nitrobenzoic acid is an easy reduction using sodium dithionite or thiourea dioxide in aq. NaOH/NH3. The water
solubility of the sodium salt will make for a rapid reduction in homogenous solution. See the other new thread about luminol prep. The reduction
methods should be quite suitable for both.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
You can easily reduce methyl m-nitrobenzoate with hydrogen over Pd-C in methanol. The yield is quantitative and the isolation could hardly be
easier. At atmospheric pressure you might need some 2 mol% of 5% Pd-C. At >3 bar <0.5 mol% Pd-C will do. I don't remember exactly if I ever
tried it on this plain methyl m-nitrobenzoate or maybe some additionally substituted methyl m-nitrobenzoate, but I'm pretty sure it works. Esterification of m-benzoic acid with methanol also gives good yields.
Using ethanol for solvent, sonicating and quenching with 3M NaOH up to pH 11 are not good ideas for your attempts with iron based reductions.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
clearly_not_atara
International Hazard
Posts: 2692
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
| Quote: | | If you give up on saving the ester, 3-nitrobenzoic acid is an easy reduction using sodium dithionite or thiourea dioxide in aq. NaOH/NH3. The water
solubility of the sodium salt will make for a rapid reduction in homogenous solution. |
You might be able to make dithionite in situ with sodium bisulfite and zinc in ethylene glycol. Sodium dithionite is usually produced by
Na2S2O5 + Zn >> Na2S2O4 + ZnO, but it is extremely flammable and thus difficult to isolate. NaHSO3 has some limited solubility in alcohols;
glycol happens to do better than most alcohols at dissolving inorganic salts.
(First, make sure that ethylene glycol will dissolve both your substrate and sodium bisulfite!)
(Yes, I searched for the solubility of Na2S2O5 in ethylene glycol; couldn't find anything definitive)
Edit: this patent seems to agree that bisulfite dissolves in Et(OH)2.
[Edited on 4-3-2016 by clearly_not_atara]
|
|
|
Archy12345
Harmless
Posts: 6
Registered: 4-3-2016
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Nicodem  |
Using ethanol for solvent, sonicating and quenching with 3M NaOH up to pH 11 are not good ideas for your attempts with iron based reductions.
|
If you read the paper I attached a link for in my original post, there has been good success with using ethanol and sonication for this sort of
reduction.
I am fairly certain my issue resides in my work up. I can't find any good instructions on how to isolate amines from their reactions mixtures for this
kind of reaction. So I'm just going off my own sep schemes. Any ideas for how to better isolate the compound?
|
|
|
Archy12345
Harmless
Posts: 6
Registered: 4-3-2016
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by UC235 | | If you give up on saving the ester, 3-nitrobenzoic acid is an easy reduction using sodium dithionite or thiourea dioxide in aq. NaOH/NH3. The water
solubility of the sodium salt will make for a rapid reduction in homogenous solution. See the other new thread about luminol prep. The reduction
methods should be quite suitable for both. |
As much as I would like to scrap the idea of keeping the ester around, it would unfortunately defeat the overall purpose of my reaction series.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Archy12345 | | If you read the paper I attached a link for in my original post, there has been good success with using ethanol and sonication for this sort of
reduction. |
I can't see methyl m-nitrobenzoate among the checked substrates. You must have misread something.
I seriously doubt that any serious chemist would use ethanol as a solvent for a methyl ester, except under neutral and mild conditions and where
product purity is not an issue.
Using NaOH up to a pH where methyl and ethyl esters rapidly hydrolyze certainly does no good either. Why didn't you use NaHCO3?
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Archy12345
Harmless
Posts: 6
Registered: 4-3-2016
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Nicodem | | Quote: Originally posted by Archy12345 | | If you read the paper I attached a link for in my original post, there has been good success with using ethanol and sonication for this sort of
reduction. |
I can't see methyl m-nitrobenzoate among the checked substrates. You must have misread something.
I seriously doubt that any serious chemist would use ethanol as a solvent for a methyl ester, except under neutral and mild conditions and where
product purity is not an issue.
Using NaOH up to a pH where methyl and ethyl esters rapidly hydrolyze certainly does no good either. Why didn't you use NaHCO3?
|
First, please cut me some slack. I am still an undergrad, and this is my first time trying to do a synthesis that I can't find much on. I am here to
learn.
Second, you're right. Methyl 3-nitrobenzoate is not among the substrates they checked. I said "this sort of reduction," not "this reduction." Among
their substrates they did have an ester compound which gave good reduction yields using their general procedure. I hadn't realized that my compound
being a methyl ester made that big of a difference, but now that I think about it I can definitely see how it would make a big difference. This is why
I didn't think of using sodium bicarbonate.
Also, just how basic does normally does the aqueous phase have to be to effectively extract an amine out?
Third, is the reason ethanol is a bad solvent in these conditions because of the likelihood of transesterification? Which solvent would you suggest
switching it out with?
|
|
|
DraconicAcid
International Hazard
Posts: 4278
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by Archy12345 | Also, just how basic does normally does the aqueous phase have to be to effectively extract an amine out?
Third, is the reason ethanol is a bad solvent in these conditions because of the likelihood of transesterification? Which solvent would you suggest
switching it out with? |
If it's a methyl ester, use methanol.
The pKa of regular ammonium salts tends to be around 9-10, so a bit more basic than that. Aniline and its derivatives tend to be a lot less basic-
the pKb of aniline is 9.1, so it could be extracted above a pH of 5. An ester of aminobenzoic acid should be similar.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Archy12345
Harmless
Posts: 6
Registered: 4-3-2016
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DraconicAcid | | Quote: Originally posted by Archy12345 | Also, just how basic does normally does the aqueous phase have to be to effectively extract an amine out?
Third, is the reason ethanol is a bad solvent in these conditions because of the likelihood of transesterification? Which solvent would you suggest
switching it out with? |
If it's a methyl ester, use methanol.
The pKa of regular ammonium salts tends to be around 9-10, so a bit more basic than that. Aniline and its derivatives tend to be a lot less basic-
the pKb of aniline is 9.1, so it could be extracted above a pH of 5. An ester of aminobenzoic acid should be similar. |
Well, I guess the methanol answer should have been right in my face.
I completely forgot that it's possible to infer a good pH for extraction based on basicity constants. Thank you for reminding me.
And if I'm thinking through this correctly, the electron withdrawing nature of the ester group should actually even raise the pkb of the amine a
little bit, right?
Perhaps if I just bring the solution to neutral pH would get good results. At 7 I am really slowing down hydrolysis kinetics, I have the vast majority
of iron out of solution. And I should be able to extract the amine.
Thank you everyone for all of the input! I will take all of these things into consideration tonight when I run this reaction yet again.
|
|
|
chemplayer..
Awesome
Posts: 48
Registered: 12-2-2016
Member Is Offline
Mood: No Mood
|
|
We did a trial of interesting nitro-reduction methods in a video a while back. The one which worked best was sodium dithionite using sodium carbonate
as a base (made fresh from sodium bisulfite and zinc powder).
We actually used this process in a follow up to produce methyl 4-aminobenzoate (benzocaine) so it seems to be relatively ester-friendly, but the
yields weren't spectacular, so there was probably some degree of hydrolysis occurring.
|
|
|
DraconicAcid
International Hazard
Posts: 4278
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
I wonder....if the ester was hydrolyzed, would the iron salt be insoluble? I know a lot of anthranilates are, but iron(III) doesn't form a simple
anthranilate.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Archy12345
Harmless
Posts: 6
Registered: 4-3-2016
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DraconicAcid | | I wonder....if the ester was hydrolyzed, would the iron salt be insoluble? I know a lot of anthranilates are, but iron(III) doesn't form a simple
anthranilate. |
I'm not sure. I imagine that most of the iron would be precipitating out as a hydroxide though because the concentration of hydroxide is so much
higher than the amine.
The other thing i was just thinking about is I might decide to not add water into my reaction mixture. That just seems like a bad idea with the methyl
ester. Perhaps instead of using 2/2/1 Methanol/Acetic Acid/Water I should use 2/3 Methanol Acetic Acid. I guess the presence of water gives the iron
ions somewhere to go, but will removing water hinder the reaction? I imagine methanol absorbs enough water out of the air (East Coast) to solvate
whatever needs solvating.
[Edited on 5-3-2016 by Archy12345]
|
|
|
chemplayer..
Awesome
Posts: 48
Registered: 12-2-2016
Member Is Offline
Mood: No Mood
|
|
Would be worth trying with copper (II). Adding copper sulfate solution to the filtrate after doing an anthranilic acid synth via Hoffman rearrangement
gets you a fair bit of copper anthranilate precipitate. The only trouble then is how to convert to the acid without something like H2S (highly toxic).
|
|
|
DraconicAcid
International Hazard
Posts: 4278
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
Anyone know if this turned out? I'm working on exactly the same reaction....
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Metacelsus
International Hazard
Posts: 2531
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
When I was making p-aminobenzoic acid, I had much better results using tin granules instead of iron (both with HCl). That might be worth a try.
|
|
|









{kind=link}