| Pages:
1
2
3
..
5 |
Melgar
Anti-Spam Agent

Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
Synthesis of dopamine from catechol
Since dopamine is often referred to as a "catecholamine", I thought it would be interesting to make an educational video where catechol is converted
to dopamine. Researching this subject has led me to conclude that the simplest way to do this would be to convert it to a benzaldehyde, then a
nitroaldol reaction with nitromethane, then perhaps a reduction with zinc. Even though I have reagents like cyanide salts and sodium borohydride, I'd
rather avoid using them, because sodium cyanide is quite toxic and hard for most people to get, and sodium borohydride feels like cheating.
One thing that I've noticed is that aromatic formylation reactions that are ortho-directing with phenol, seem to be para-directing with catechol. I don't know if this is typically
the case, or only true of some reactions. If it's true of the Duff reaction, that might be the way to go:
https://en.wikipedia.org/wiki/Duff_reaction
Now, I've been recommended that it might be better to form benzodioxole first to protect the fairly reactive phenol groups. That seemed a little
sketchy at first, but I guess benzaldehyde is a lot easier to get than piperonal, and if someone is able to source nitroethane already, they're better
off reacting it with piperonal than benzaldehyde. Harm reduction! 
Also, I smelled piperonal a long time ago, and it was amazing. Like maraschino cherries, and very strong. So that would be a good indication of
success at that step.
Another option might be to just cleave a methyl or ethyl group off of vanillin/ethylvanillin, which I'd do instead if it was a lot less work and
better yields. I probably would do that anyway if it was practical, just to compare different routes and make sure I was on the right track.
Any thoughts?
As an aside, the secret motive behind a portion of the video series that we're planning is to lure in the Walter White wannabes, then make them learn
a whole bunch of background chemistry and biology. We had another idea to show faked reactions that look really neat and clean, but are actually some
of the most horrible glassware-ruining reactions that exist; think pyrolysis of polystyrene to get styrene, for example. Ultimately, teaching them
science seemed like a more noble motive, and besides, how many of us now have a legitimate interest in chemistry that started because we wanted to
make explosives or idolized Shulgin?
The first step in the process of learning something is admitting that you don't know it already.
I'm givin' the spam shields max power at full warp, but they just dinna have the power! We're gonna have to evacuate to new forum software!
|
|
|
Tsjerk
International Hazard
Posts: 3022
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I think your secrete motive might actually work for one or two smart guys out there
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
you can make protocatechualdehyde from catechol using the reimer tiemann reaction - Reimer and Tiemann, Ber. 9, 1268 (1876); Tiemann and Koppe, ibid.
14, 2015 (1881).
from there,dopamine is 2 steps away
EDIT- In that case,you could go balls to the wall and do a fenton on tyrosine to get DOPA and then decarboxylate that to get dopamine -https://www.ncbi.nlm.nih.gov/pubmed/1321588
or even better,directly hydroxylate tyramine -http://onlinelibrary.wiley.com/doi/10.1002/chem.200500361/fu...(they use a protein but you could try using just
H2O2/NO2-  ) )
[Edited on 23-10-2017 by CuReUS]
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
Quote: Originally posted by CuReUS  | you can make protocatechualdehyde from catechol using the reimer tiemann reaction - Reimer and Tiemann, Ber. 9, 1268 (1876); Tiemann and Koppe, ibid.
14, 2015 (1881).
from there,dopamine is 2 steps away |
Have you ever done this, or attempted it? I have, it's an enormous mess. It's a free-radical reaction, and the yields will necessarily be very low,
after the nightmarish workup.
Okay, I exaggerated a little. But suffice to say the yield certainly wasn't worth the effort of the workup.
The first step in the process of learning something is admitting that you don't know it already.
I'm givin' the spam shields max power at full warp, but they just dinna have the power! We're gonna have to evacuate to new forum software!
|
|
|
Corrosive Joeseph
National Hazard
Posts: 915
Registered: 17-5-2015
Location: The Other Place
Member Is Offline
Mood: Cyclic
|
|
Excellent choice of research.......... A most fascinating compound indeed
And an interesting choice of routes and precursors to boot
/CJ
[Edited on 22-10-2017 by Corrosive Joeseph]
|
|
|
SWIM
National Hazard
Posts: 970
Registered: 3-9-2017
Member Is Offline
|
|
Have you considered going the natural route?
Extract L-dopa from broad beans and use your considerable experience with decarboxylations to make dopamine.
You could even do this as well as your other synthesis and then you'd be able to confirm their equivalence with a mixed melting point test.
As for the vanillin demethylation Idea there's a high yielding reaction that uses a 10% excess of aluminum chloride suspended in a methylene chloride
solution of vanillin which then has pyridine added (4.4 equivalents) under stirring and is refluxed for 48 hrs to give a claimed 87% yield.
Robert G Lange, J. Org, 27, 2037 (1962)
I haven't looked up the journal entry as I'm pretty sure I'd have to pay through the nose for it, but it is summarized in Feiser and Feiser's Topics
in Organic Chemistry.
Actually, I suppose L-dopa might be a little more heat sensitive than tryptophan so maybe that 1st Idea is a non-starter unless you protect those OH
groups.
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Melgar | | Quote: Originally posted by CuReUS | you can make protocatechualdehyde from catechol using the reimer tiemann reaction - Reimer and Tiemann, Ber. 9, 1268 (1876); Tiemann and Koppe, ibid.
14, 2015 (1881).
from there,dopamine is 2 steps away |
Have you ever done this, or attempted it? I have, it's an enormous mess. It's a free-radical reaction, and the yields will necessarily be very low,
after the nightmarish workup.
Okay, I exaggerated a little. But suffice to say the yield certainly wasn't worth the effort of the workup. |
Small correction: the Riemer-Tiemann is not a free-radical reaction. It is a simple electrophillic aromatic substitution reaction by an in-situ
generated dichlorocarbene species. First, chloroform is deprotonated to the trichloromethylide anion, and this then loses a chloride to become
reactive dichlorocarbene. This carbene is very electron-poor, so it will attack the nearest source of electron density it can find, which happens to
be the electron-rich ortho and para positions of a phenol. It replaces the proton which lies on the ring, the proton attaches itself to the carbene,
and the chlorines are replaced with water to form the final formyl group.
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
If you really REALLY wanted to go balls to the wall and were still contempt on the catechol route then you could extend the series to also cover the
synthesis of catechol from the somewhat OTC naturally occurring salicylic acid.
By converting it to salicylamide and then hoffman degradation to 2-aminophenol and then diazotize the amine and finally hydrolize the diazonium anion
in hot copper sulfate solution.
http://www.sciencemadness.org/talk/viewthread.php?tid=63327
I have done this and most of it is close to quantitative except the Hoffman which produced 80% yield if i recall correctly.
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
| Quote: Originally posted by Cryolite. | | Small correction: the Riemer-Tiemann is not a free-radical reaction. It is a simple electrophillic aromatic substitution reaction by an in-situ
generated dichlorocarbene species. First, chloroform is deprotonated to the trichloromethylide anion, and this then loses a chloride to become
reactive dichlorocarbene. This carbene is very electron-poor, so it will attack the nearest source of electron density it can find, which happens to
be the electron-rich ortho and para positions of a phenol. It replaces the proton which lies on the ring, the proton attaches itself to the carbene,
and the chlorines are replaced with water to form the final formyl group. |
And isn't dichlorocarbene a free radical, with two vacancies? I guess another way to say it is that it is very electron poor, but the whole reason
for that is that it's a free radical, no?
I wanted to do something that isn't going to require things that most people can only get on rare occasions, when eBay is looking the other way. I
like vanillin because of how easy it is to get, although I kind of wanted to start with catechol. I might just explain that catechol is a component
of coal tar and creosote, and that because of how bad that is for you, it's hard to get anymore. It's so common in creosote though, that it seems a
bit masochistic to go through too much effort to make it.
The vanillin demethylation method that was mentioned earlier seems to be legitimate, although the pyridine part could be a sticking point. I read
somewhere else that the bond disassociation energy is lowest for that particular methyl group, meaning that free radical temperatures with reducing
conditions could do it too, if conditions were carefully controlled.
What about the Duff reaction though? Anyone have any experience with that?
The first step in the process of learning something is admitting that you don't know it already.
I'm givin' the spam shields max power at full warp, but they just dinna have the power! We're gonna have to evacuate to new forum software!
|
|
|
clearly_not_atara
International Hazard
Posts: 2691
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
You think you could just alkylate catechol with nitroethylene directly and save a few steps? There are a few systems I know that carry out this rxn,
eg:
http://onlinelibrary.wiley.com/doi/10.1002/ejoc.201300579/ab...
http://onlinelibrary.wiley.com/doi/10.1002/ejoc.201000271/fu...
http://www.sciencedirect.com/science/article/pii/S0021967300...
http://www.sciencedirect.com/science/article/pii/S0040403901...
Although an OTC procedure hasn't popped up I suspect that Bronsted acid catalyst shouldn't be too hard to manage if you pick the right one.
[Edited on 23-10-2017 by clearly_not_atara]
[Edited on 23-10-2017 by clearly_not_atara]
[Edited on 23-10-2017 by clearly_not_atara]
[Edited on 04-20-1969 by clearly_not_atara]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
I think making catechol from aspirin looks like fun, but somehow I doubt the yields would be as good as they look on paper. Then again, aspirin is
pretty cheap. I really need to finish working up the toxic mess sitting in my fume hood so I can do some other experiments....
|
|
|
chemplayer...
Hazard to Others
Posts: 191
Registered: 25-4-2016
Location: Away from the secret island
Member Is Offline
Mood: No Mood
|
|
Dopamine was on our wish-list of cool things to make (and do an educational video on), so here's a summary of our journey on various paths:
We tried about 10 times with different ways and different conditions to demethylate vanillin, and with limited success and very poor yields. This is
not easy! We make AlCl3 home-made from ZnCl2 however so maybe the zinc impurity interferes with the reaction.
We did successfully make piperonal from peppercorns (extraction, hydrolysis, then oxidation). Not great yields, but enough to play with (gram
quantities are feasible), so that would open up the nitromethane route.
Another possible route was to prepare the vanillin-related compound first and then try to demethylate. Vanillin nitro-methane adduct we did produce
and it's easy to make and purify. We reduced the nitrostyrene to the nitroalkane but didn't go any further. Can't remember why we didn't take this
another step - not sure if we had any reducing agent capable of going that final step, or if we just thought it would be too controversial a reaction
to do a video on (even though the product is not pharmacologically active).
We also wanted to try to make vanillyl chloride and then nucleophilically react with cyanide (plus a reduction), but vanillyl chloride turned out to
be a lot harder than it looks to make (HCl + zinc chloride + vanillin seem to form a purple complex - again, zinc gets in the way)...
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
where have you noticed this ?
truly going BTW would be doing a one pot schmidt/diazotisation on aspirin.It would first get converted to acetyl aminophenol,then NaNO2
could be added which would react with the excess H2SO4 and diazotise the amine.Boiling this solution would convert the diazonium
to OH and de-acetylate at the same time to give catechol.
| Quote: Originally posted by Melgar |
And isn't dichlorocarbene a free radical, with two vacancies? I guess another way to say it is that it is very electron poor, but the whole reason
for that is that it's a free radical, no? |
this is quite interesting actually.Triplet carbenes are considered free radicals but not singlet carbenes.Which of the two is :CCl2 ?
| Quote: | | What about the Duff reaction though? Anyone have any experience with that? |
I think a duff reaction on catechol would give benzodioxole instead of the desired product.Even if it worked,the formylation would be ortho and there
might be double formylation as well | Quote: Originally posted by clearly_not_atara | | You think you could just alkylate catechol with nitroethylene directly and save a few steps? Although an OTC procedure hasn't popped up I suspect that
Bronsted acid catalyst shouldn't be too hard to manage if you pick the right one. |
That's pure genius  . Do you think PPA or ZnCl2 could be used ? . Do you think PPA or ZnCl2 could be used ?
[Edited on 24-10-2017 by CuReUS]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
Say, I was doing some reading on borohydride earlier, and Vogel Practical Organic Chemistry 3rd edition says that it won't reduce nitro groups or
alkenes... will it reduce a nitrostyrene?
|
|
|
Chemi Pharma
Hazard to Others
Posts: 349
Registered: 5-5-2016
Location: Latin America
Member Is Offline
Mood: Quarantined
|
|
| Quote: Originally posted by JJay | | Say, I was doing some reading on borohydride earlier, and Vogel Practical Organic Chemistry 3rd edition says that it won't reduce nitro groups or
alkenes... will it reduce a nitrostyrene? |
If you use borohydride with nickel salt, such as nickel chloride you will produce nickel boride that's capable of easy reduct the nitro group and the
double bond.
See: http://www.sciencemadness.org/talk/viewthread.php?tid=66278&...
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
| Quote: Originally posted by JJay | | Say, I was doing some reading on borohydride earlier, and Vogel Practical Organic Chemistry 3rd edition says that it won't reduce nitro groups or
alkenes... will it reduce a nitrostyrene? |
It'll reduce it to a nitroalkane. Because the double bond is highly polarized, NaBH4 can actually reduce it, which is a fairly common thing to do,
since nitroalkanes reduce easier, and with the double bond reduced, they won't react with each other during the nitro reduction stage.
The first step in the process of learning something is admitting that you don't know it already.
I'm givin' the spam shields max power at full warp, but they just dinna have the power! We're gonna have to evacuate to new forum software!
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
Hmm... I meant to buy some nickel chloride a while back and never got around to it. That will definitely have to go on the list.
I just read about a borane-THF adduct capable of reducing carboxylic acids to alcohols. There are papers that describe making it with sodium
borohydride and iodine in THF.
|
|
|
Chemi Pharma
Hazard to Others
Posts: 349
Registered: 5-5-2016
Location: Latin America
Member Is Offline
Mood: Quarantined
|
|
| Quote: Originally posted by Melgar | | Quote: Originally posted by JJay | | Say, I was doing some reading on borohydride earlier, and Vogel Practical Organic Chemistry 3rd edition says that it won't reduce nitro groups or
alkenes... will it reduce a nitrostyrene? |
It'll reduce it to a nitroalkane. Because the double bond is highly polarized, NaBH4 can actually reduce it, which is a fairly common thing to do,
since nitroalkanes reduce easier, and with the double bond reduced, they won't react with each other during the nitro reduction stage.
|
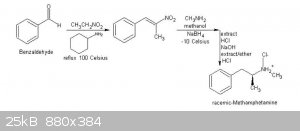
By the way @Melgar, could you explain to me why sodium borohydride alone can reduce phenyl nitropropene with methylamine directly to metamphetamine in
a kind of reductive amination?
I read about an experiment like this on this book:
A Laboratory History of Narcotics
Vol. 1: Amphetamines and derivatives
A Laboratory Manual - 2007 - page nº 111
Jared B. Ledgard
But I can't explain why this can go on, since I ever thought borohydrides alone only reduce the double bond of the alkene to an alkane, but not the
nitro group.
Could you or someone here explain to me the mechanisms envolved in this reaction below showed on that book? Is it possible?
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
@ChemiPharma Sodium borohydride can only reduce double bonds that are sufficiently polarized by the groups on either side of it. For most alkenes,
this isn't the case, but if, say, there's a strongly electron-withdrawing group on one side and not the other, that will be enough for borohydride to
be able to align itself right to reduce it. Sodium borohydride can reduce imines for the same reason it can reduce aldehydes and ketones: because
nitrogen (like oxygen) is more electronegative than carbon, and sodium borohydride is good at reducing double bonds to carbon when the carbon is
double-bonded to something more electronegative than itself.
Sort of a simplified explanation, but it helps to realize that when sodium borohydride reduces double bonds in imines and nitroalkenes, it's really
the same mechanism as when it reduces aldehydes and ketones.
Oh, also, you'll have to quote someone more authoritative than that notorious idiot if you want anyone here to believe that the reaction you describe
is real.
[Edited on 10/24/17 by Melgar]
The first step in the process of learning something is admitting that you don't know it already.
I'm givin' the spam shields max power at full warp, but they just dinna have the power! We're gonna have to evacuate to new forum software!
|
|
|
myristicinaldehyde
Hazard to Others
Posts: 166
Registered: 23-4-2016
Location: .͐͌ ͛҉̻̫̰̻̖E̮ͮ̐́̚ ̢̗̅̉ͩ͂̒̌.̯̻̺̯̀̎͂̄ͩ̚
Member Is Offline
Mood: сорок пять
|
|
This is an interesting alternative to dopamine, via a pummerer rearrangement and subsequent reduction.
https://www.jstage.jst.go.jp/article/cpb1958/37/12/37_12_339...
The only problem might be preparing the needed methylsulfinylacetamide.
If we don't study the mistakes of the future we're doomed to repeat them for the first time.
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
The THF-borane adduct will reduce nitro groups, but I think it hydroborates styrenes. So I suppose it could be used for anti-Markovnikov hydration of
some styrenes, but I am not exactly sure what would happen here.
|
|
|
Chemi Pharma
Hazard to Others
Posts: 349
Registered: 5-5-2016
Location: Latin America
Member Is Offline
Mood: Quarantined
|
|
| Quote: Originally posted by Melgar | | Oh, also, you'll have to quote someone more authoritative than that notorious idiot if you want anyone here to believe that the reaction you describe
is real. |
Sorry @Melgar, but I didn't understand the last part.
This reaction was extracted from a published book.
Do You believe that's impossible to occurs? The author is a fraud? yes or not? why?
Here's the complete text at the book I have cited, on pages 111/113:
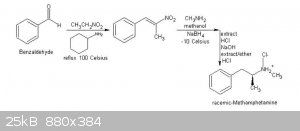
Procedure C: Preparation of racemic-methamphetamine hydrochloride (ICE)
Materials•
I. 27 grams of benzaldehyde 8. 30 grams of anhydrous sodium sulfate
2. 20 grams ofnitroethane 9. 3.75 grams of sodium borohydride
3. 5 milliliters of cyclohexylamine 10. 200 mi IIi I iters of methylene chloride
4. 300 milliliters of dry hexane 11. 60 grams of dry hydrogen chloride gas
5. 500 milliliters of dry methanol 12. 50 grams of sodium hydroxide
6. 25 grams of methylamine gas 13. 450 milliliters of diethyl ether
7. 55 grams of anhydrous magnesium sulfate
Summary: racemic-Methamphetmaine (ice), is prepared by reacting phenyl -2-nitropropene with methylamine and sodium borohydride in the presence of
methanol. The phenyl-2-nitropropene is formed by the condensation of benzaldehyde with nitroethane in the presence of cyclohexylamine. After
the reaction of phenyl-2-nitropropene with sodium borohydride and methanol , the reaction mixture is treated with methylene chloride, and this
resulting methylene chloride mixture is then treated with hydrogen chloride, and the resulting acidified mixture is then evaporated to remove
the bulk of the methylene chloride. After the bulk of the methylene chloride has been removed, the left over remaining concentrate is then
filtered to recover the precipitated product. The precipitated product is then treated with sodium hydroxide to liberate the freebase, which
is then extracted into ether. The ether extract is then treated with hydrogen chloride to precipitate the ICE, which is then filtered-off
and then vacuum dried or air-dried.
Step I: Preparation of phenyl-2-nitropropene
Into a suitable reflux apparatus, place all at once, 27 grams of benzaldehyde, followed by 20 grams of nitroethane, followed by 5 milliliters of
cyclohexylamine. Thereafter, reflux the entire mixture at about 100 Celsius for 3 hours. After refluxing for 3 hours, remove the heat source, and
allow the two-phase reaction mixture to cool to room temperature. Then pour the entire reaction mixture into a seperatory funnel , and remove
the lower (organic) layer. The upper layer can be recycled or discarded if desired as it will contain the cyclohexylamine catalyst. Then place the
recovered lower organic layer into a suitable sized beaker, and then add in 25 milliliters of cold water. Immediately thereafter, rapidly stir
the mixture using magnetic stirrer, or other means, for about 30 minutes at room temperature. Then remove the upper water layer by decanting it off,
and then place the lower organic layer (containing the desired product), into an ice bath and chi ll to about 0 Celsius. Then, add in about 10
milliliters of cold water, and then allow the total mixture to stand at room temperature for several hours to allow the desired product of
phenyl-2-nitropropene to crystal! ize. After 2 hours, most of the desired nitro compound should have precipitated and afterwards, filter-off the
precipitated crystals, and then vacuum dry or air-dry them. Finally, recrystallize these dried collected crystals from 150 milliliters of dry
hexane, and after the recrystallization process, vacuum dry or air-dry the crystals.
Step 2: Preparation of racemic-methamphetamine
Into a suitable beaker, place 250 milliliters of dry methanol, and then bubble into this methanol , 25 grams of methylamine gas. After the addition
of the methylamine gas, add to the resulting methylamine/methanol solution, 25 grams of anhydrous magnesium sulfate (to absorb water), and then
place this entire mixture into a suitable flask , and then stopper the flask .
Immediately thereafter, stir the entire mixture for about 10 minutes. After I 0 minutes, carefully filter-off the magnesium sulfate, and do it
as fast as possible to avoid moisture absorption by the methylamine/methanol mixture. After the filtration,
place the methylamine/methanol mixture into a suitable 3-neck flask (equipped with motorized stirrer, thermometer, and addition funnel), and then
place a phenyl-2-nitropropene solution into the addition funnel-this solution being prepared by adding and dissolving 34 grams of
phenyl-2-nitropropene (obtained in step I) into 150 milliliters of dry hexane. Note: the 3-neck reaction flask should be equipped with a
calcium chloride drying tube to keep moisture from entering the apparatus. Then
place the 3-neck reaction flask into a cold-water bath at about I 0 to 15 Celsius. Then add to the methylamine/methanol mixture, 30 grams of
anhydrous sodium sulfate. Note: this sodium sulfate is to absorb any water formed during the reaction. Note: dried silica gel pieces can be used
instead of sodium sulfate if desired. Now, slowly add drop-wise, the phenyl-2- nitropropene/hexane solution form the addition funnel , to the
methylamine/methanol mixture over a period sufficient to keep the reaction mixture below 25 Celsius at all times. During the addition, moderately
stir the reaction mixture with the motorized stirrer. After the addition of the nitropropene/hexane solution, continue to stir the reaction
mixture for I hour at a temperature below 25 Celsius. After this additional I hour of mixing, stop stirring, and then quickly filter the
reaction mixture to remove the insoluble sodium sulfate. Then place this filtered reaction into a clean flask, and then place this flask into
an ice bath, and chill to - I 0 Celsius. Note: while waiting for the reaction mixture to chill, stopper the flask to keep moisture out. When
the temperature of the reaction mixture reaches - I 0 Celsius, remove the stopper, and replace it with a standard powder funnel , and then slowly
add in, in small portions at a time, 3.75 grams of sodium borohydride, and after each portion, add in 25 milliliters of methanol (ten
375-milligram portions of sodium borohydride and ten 25-milliliter portions of methanol). During the entire addition, rapidly stir the reaction
mixture, and maintain its temperature below 20 Celsius at all times. After the addition of the sodium borohydride and methanol portions, continue
to stir the entire reaction mixture at a temperature below 20 Celsius for about 3 hours. After 3 hours, pour the entire reaction mixture into a
large flask, and then add in 1200 milliliters of water. Shortly thereafter, add in 200 milliliters of methylene chloride, and then rapidly
stir the reaction mixture for about 30 minutes at room temperature. Then decant-off (pour-off) the upper aqueous layer, and then place the
remaining lower methylene chloride layer into a seperatory funnel , and drain-off the lower methylene chloride layer- as there will be some
upper aqueous layer still remaining. Thereafter, place the methylene chloride layer into a beaker, and then add in 15 grams of anhydrous
magnesium sulfate (to absorb water) . Then stir the entire mixture for about 10 minutes, and then filter-off the magnesium sulfate. Then, place
the entire mixture into a suitable sized beaker, and then bubble into the mixture, 30 grams (excess) of dry hydrogen chloride gas. After the
addition of the hydrogen chloride gas, place the entire acidified mixture into a distillation apparatus or rotary evaporator, and distill-off the
methylene chloride at 40 Celsius until only 80% of the total volume remains. Once this point is reached, stop the distillation process, and then
remove the left over remaining contents (after it has cooled to room temperature), and then filter these contents to recover the precipitated
impure product. Then vacuum dry or air-dry the filtered-off crystals. Thereafter, place these crystals into a clean beaker, and then add in a
sodium hydroxide solution prepared by adding and dissolving 50 grams of sodium hydroxide into 250 milliliters of water, and after the addition, stir
the entire three 150-millilter portions of diethyl ether, and after the extraction, combine all ether portions (if not already done so), and then
dry this combined ether portion by adding to it , 15 grams of anhydrous magnesium sulfate. Then stir the entire mixture for mixture for about 30
minutes at room temperature. Note: sodium hydroxide generates much heart when dissolved in water, so allow the solution to cool before using. After
stirring the alkaline mixture for about 30 minutes, extract the entire mixture with with three 150-millilter portions of diethyl ether, and after
the extraction, combine all ether portions (if not already done so), and then dry this combined ether portion by adding to it , 15 grams of
anhydrous magnesium sulfate. Then stir the entire mixture for about 10 minutes, and then filter-off the magnesium sulfate. Finally, place the
filtered ether mixture into an ice bath, and chill to 0 Celsius. Thereafter, bubble into the ether mixture, 30 grams of dry hydrogen chloride
gas (excess), and after the addition, stir the entire ether mixture for about 30 minutes. Thereafter, filter-off the precipitated crystals, and
then vacuum dry or air-dry the crystals.
[Edited on 24-10-2017 by Chemi Pharma]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
No yields. Gas measurements don't look very practical. I mean, sure, you could weigh your methylamine and hydrogen chloride cylinders while in use,
but you would need a high-capacity scale. And you could also weigh the methanol solution, but that won't work for the hydrogen chloride. I don't think
these procedures were tested. Also, the link to dopamine synthesis here is rather sketchy.
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
you could do a fries rearrangement on catechol with chloro acetylchloride to get 3,4-dioxychloracetophenone.then replace the Cl with NH2
and reduce the ketone to get dopamine
I wonder if glycine could be used directly instead of chloro acetylchloride.

|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
This is the first time I have heard of the Fries rearrangement... interesting. Why doesn't the chloro acetylchloride react with pyridine?
|
|
|
| Pages:
1
2
3
..
5 |
|








