Pumukli
National Hazard

Posts: 686
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
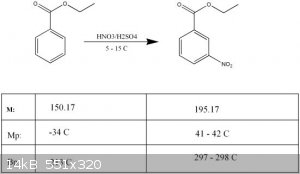
Synthesis of m-nitro ethylbenzoate
Synthesis of m-nitro ethylbenzoate
Literature: Orgsynth, Vol. 3, p. 71 (1923)
Reaction data:
Daily annoyance:
I started putting together the required equipment for this synthesis when I realized that I can't find my one and only ptfe stirbar. The original
Orgsynth article called for a mechanical stirrer, though in 1923 I think they rarely used magnetic stirrers and I thought I'd try with magnetic
stirring anyway. (I don't have mechanical stirring capability at the moment so the route was given.) I checked and rechecked every possible storage
place in my lab several times but to no avail. I got very frustrated because I had the time to do the synthesis but the reaction is sensitive to the
temperature so stirring is a must.
I decided I'd cobble together a home-made stirbar. I had an old hard disk magnet, broke it into smaller pieces (it was brittle), wrapped a
good-looking piece into polyethylene bag cuttings and with a heat gun I tried to heatseal it in PE all around. It quite worked and got something that
was remotely similar to a real stir bar. Unfortunately it was not a great performer. Main problem was that above a certain rpm of the stirrer the bar
changed its spinning characteristics: was not spinning perpendicular to its long axis anymore but started spinning parallel with it! It spinned like
crazy but moved hardly any liquid. :-)
Synthesis description:
1/6th batch size of the original description.

Into a 250 ml round bottom flask I measured 75 milliliters (approx. 1.4 mol) H2SO4 and put the homemade stir bar in, then cooled the contents of the
flask to around 7 Celsius with the help of snow/ice/water bath.
When the acid was cooled I added 38.5 g (0.256 mol) ethylbenzoate into it all at once. The temperature rose to 30 Celsius immediately then the mixture
slowly cooled down. (Stirrer was spinning.) It formed a clear liquid. When the temperature went below 10 C I started slowly dropping in the nitrating
acid mixture from a dropping funnel. This mixed acid was made of 21 ml cc. HNO3 and 15 ml cc. H2SO4. (A note to the beginner: when you make such mixed
acid it heats up considerably! Be prepared for cooling it!)
The dropping was very slow, but this way the temperature of the mixture never rose above 10 Celsius. It took me more than 3 hours with the weak
homemade stir bar to do this. The last 20% of the nitrating mix could be dripped in more quickly than the preceding 80% because it causes much lower
temperature rise. I stirred and cooled the mixture for an additional 20 minutes after I finished the introduction of the mixed acid.

During this 20 minutes I made 200 g snow/fine ice mixture in a jar and after the 20 minutes extra reaction time I poured on the content of the flask
on the snow/ice mixture. The snow melt immediately but kept the whole liquid cool. At first the crude product separated as a yellow oil but quickly
turned to whitish solid as I stirred it with the thermometer. (Bad habit I know, don't follow, use a glass rod instead!) It formed a waxy precipitate
which attached itself strongly to the glass thermometer. At this point I noted that to my surprise I could not feel the strong ethylbenzoate odour at
all! I expected the mixture would be smelly from a few percent of unreacted benzoate at least but no, there was not that smell at all!

I filtered this diluted acidic mixture on a Buchner funnel with filter paper. It worked. I was a bit wary at first because I thought the acid might
dissolve the filter paper as well but apparently at this concentration it did not.

Then I started pouring ice cold water on it and started to „kneading” it in the Buchner. The original article did not mention it but when I
washed with water it quickly turned into a white gummy mass as if I was trying to wash a sizeable blob of soft chewing gum! I was actually kneading it
with a glass rod!
What I got crude gummy solid:

I put into a 100 ml flask and triturated with 35 ml cold methanol, then filtered, then repeated this trituration/washing with another 30 ml or so
methanol. In methanol the gummy mass disintegrated at once and got a crystalline form! I filtered off the washing methanol, it was yellow, while the
crystalline mass on the filter was white with a hint of green. After 12 hours of drying the mass of the crude product was 28.6 g, its color remained
very faintly green white and had a pleasant, subtle, citrus-like odour.
The yield of the crude product was 65% which was much less than expected so I repeated the whole synthesis again, with the exact same ammounts of
reagents. Somehow I felt that the insufficient stirring with the homemade stir bar and the too sloow addition of the nitrating mix and the too long
reaction time may caused more side reactions and lowered the yield.
I decided I would allow higher reaction temperatures this time because it would allow me quicker dropping rate and shorter overall reaction time. Then
unexpectedly, around mid-reaction, I found my real ptfe stirbar and put it into the mixture. It made possible a more efficient stirring and really
helped to quickly finish the reaction. The reaction temperature never excedeed 15 Celsius either! I finished the reaction in 130 minutes – which was
much quicker than the previous one (210 minutes) but was till much slower, than could possibly be done (around 60 minutes, suggestion in orgsyn).
The work up was also the same. The mass of crude product after 12 hours of drying (after the methanol wash) was 34 g. What an improvement!

Then I noticed that crystals formed in the (saved) methanol washings. I filtered, dried and weighed them. They looked like the crude product.
I mixed all crudes at this point, I got 68.5 g. Then I recrystallized it from hot methanol (40.5 g). The original article suggested using the same
ammount of methanol for recrystallization but I found that about 2/3rd was enough to dissolve everything! I filtered the warm methanolic solution
through filter paper too, it caught a lot of sand and dust and inorganic dirt. (Which was introduced with the cold water (melt snow) washings.)
After everything was finished I got 62.4g dried product.:

Its odour is very faint and pleasant, not citrus-like as before. Its colour is white with a very faint yellowish-green tint.
Final yield is 62.4% (0.3197 mol), combined crude yield was 68.5%, crude yield from the second synthesis attempt: 72.0%. I think crude yield 75%+
would be achievable with better stirring (= quicker overall reaction) and careful work-up.
I also think if I recrystallized it again then it would become true snow-white.
Finally I could make my melting point instrument working.
The mp of the recrystallized product is 38.0 - 38.5 C, which seems to be in good agreement with the fact, that it is not entirely pure. (Melting point
should be 41-42 C.)
A second recrystallization may solve it, altough I think it is satisfactorily pure for my needs.
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Nice write-up Pumikli and carried out at a reasonable preparatory scale too.
I suspect that the reason the original authors used the slightly larger quantity of solvent for the recrystallization was to obtain the higher melting
point in one go but unless you need the higher purity it may be better to go for the higher yield, particularly for amateurs where cost of reagents is
an issue.
Just out of a matter of interest did you prepare the ethyl benzoate too?
|
|
|
Pumukli
National Hazard
Posts: 686
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
I tried to save on methanol (which is a "non-renewable" for me) but in the end it might have been better to sacrifice those 20-30 extra millilitres
and get a cleaner product. :-)
Yes, I synthetized ethyl-benzoate too, the synthesis was published a few days ago in Prebublication too.
|
|
|
|








