Fleaker
International Hazard

Posts: 1252
Registered: 19-6-2005
Member Is Offline
Mood: nucleophilic
|
|
Precious Metals Reclamation Thread (Photo Intensive!!)
It's my purpose to deal with the aqueous chemistry, treatment and techniques with respect to the recovery of Au, Pd, Pt, Re, Rh, Ru, Os, Ir, and Ag.
First up are Pd, and Pt. Re will follow shortly, and hopefully Os and Ru after that.
Now for the post:
Not too long ago I did some PGM refining for a well-known member here who did not have the available time to do it himself.
Most of the material he sent was very pure, but it was in various forms of large chunks or pieces that were stuck in crucibles, or were large pieces.
Other bits of material were unknown and included some platinum based printing ink, a MnSbPt sputtering target, of unknown Pt concentration. My friend
also sent about 380g of potassium perrhenate, which I was to reduce to rhenium sponge at high temperature. I did not do this in the end, because he
would rather I do the ammonium salt, which he sent me about a week ago. It is now now processed and I'll be doing a little write-up on Rhenium from
perrhenate.
I see a few questions here occasionally that deal with platinum and palladium and their chemistry amongst others of that group. What follows is a
photodocumentary of sorts that shows the process and should serve as a general guide for refining. I hope to eventually cover all of the precious
metals and offer up some ''standard techniques'' that members here can use while trying to obtain these rarities of the periodic table. Please note
I'm not saying what I did was the best or only way to go about it, just how I did it, and how it worked out for me. There are many ways to skin a cat.
These are close-ups of the starting materials that I received.You will see that most of the palladium is in graphite crucibles or in porcelain ones. I
broke off as much of the graphite as I could, and visually checked pieces for any palladium metal. You should also notice a metal crucible. It is
actually molybdenum, with a Pt liner, with a Pt button in it.
Initial masses:
Molybdenum crucible with Pt: 42.8791g
Platinum printing ink (interesting stuff, dense, but waxy): 18.4551g
Platinum chunk with stuck on graphite crucible base: 32.9368g30.2420g
MnSbPt sputtering target: 66.3767g~1.7 g
All palladium materials: 139.5161g101.1 g


Some considerations
At the outset, I decided upon a plan of action for the scrap materials. The Mo crucible would be dissolved away in nitric acid leaving behind the
Platinum button, the Pt print ink would be rinsed with solvent, dried, then dissolved in hot aqua regia, the palladium would be dissolved in aqua
regia, the sputtering target in cold aqua regia, from which I could separate the platinum powder (as a gray material on the bottom). Everything was
kept separate to avoid any possible contamination. I suspected that the Mo crucible and the Sb part of the sputtering target might be problematic, but
I'll explain later...
The large Pt chunk had the graphite removed mechanically (carefully!) with a file and grinder, then the Pt button was brought to orange heat in a
Meker burner to rid the last bit of carbon off of its surface. It was then pure enough not to bother with any further.
My aqua regia was made with ACS grade acids, 1 part of 16M HNO3 to 3.75 parts 12M HCl.
Procedure--Palladium
So, first goal was to get all the Pd values into solution. Aqua regia doesn't appreciably attack porcelain or graphite, so I simply poured aqua regia
onto the crushed up crucibles. With either nitric acid or aqua regia, one notices an immediate formation of dark brown syrup. (This is actually so
intensely yellow, it appears brown. A small drop of concentrated solution to a liter of water would give a chromate yellow colour.) Palladium is much
less noble than platinum and dissolves readily and with evolution of heat and NOx into the aqua regia. The digestion was performed in a 400mL beaker,
and poured the spent acid to a 1L Erlenmeyer flask, which I kept boiling constantly to remove NO3-. This produces a lot of nitrogen dioxide, so care
must be exercised (use a fume hood!) While all of the small bits of Pd on the walls quickly dissolved, the big buttons of Pd were relatively unfazed,
it would take many hours to dissolve them completely. Patience is needed. Below are the pictures of a few stages of dissolution.

Just starting the process, everything is a 22*C.

Loaded beaker with Pd in graphite and alumina crucibles.

Pd dissolving away in aqua regia.
After all was dissolved, it was filtered it through a fine glass frit, rinsing the frit well with dH2O. This removed all of the graphite chunks.
After the filtration, the solution is then deNOxed in the 1L Erlenmeyer flask with HCl additions. However, it was taking overlong to get rid of all
the nitric acid so a rotary evaporator at 80*C under vacuum was utilized to reduce the volume and remove nitrogen dioxide. The evaporation took about
30 minutes to remove all of the nitric and about 200mL of water. What was left was a very syrupy and extremely dense 300mL of H2PdCl6/HCl solution.

Removing nitrogen oxides, a standard procedure for aqua regia work.

Palladic acid syrup, very heavy.
All of the Pd syrup was diluted down with 1200 mL of distilled water in a 4000mL beaker, and about 800mL of distilled water was put on the boil, to
this was added about 15g of sodium chlorate, and enough ammonium chloride to make a saturated solution. The sodium chlorate serves as an oxidizer to
produce chlorine in the HCl-acidified Pd solution. (I would have just bubbled chlorine through the solution but I could not get my Cl2 lecture bottle
open.) 600mL of the hot NH4Cl-chlorate solution was added in three portions into the diluted Pd solution, which immediately evolved chlorine gas, and
thickens to almost a gelatinous consistency. It is not often that a complete precipitation of the brick-red ammonium chloropalladate(IV)/ (NH4)2PdCl6
occurs, often it must be treated twice after a filtration, the second time cooling the whole solution in an ice bath (as seen above).

Saturated ammonium chloride with 20g/L chlorate anion.

Crashing out voluminous precipitate in the cold, 6*C

Precipitate settling. Note the solution is still dark, indicating it is incomplete.
The precipitate is filtered onto a medium fast filter paper with vacuum suction and a Buchner funnel. The precipitate is washed with the ammonium
chloride solution that was not used for the precipitation. After the second precipitation of ammonium chloropalladate, the filtrate should be clear,
and the ammonium chloride rinse should also run clear. If it is not, reprecipitate and chill til it runs clear.

Vacuum filtration with Buchner, filtrate is still coloured.

Finally, all Pd values are stripped from solution.

Ammonium hexachloropalladate precipitate in the funnel.
The dry precipitate is shown --all of the filter papers and precipitate were put in a vacuum dessicator and left until dry. A razor blade was used to
remove as much precipitate off of the filter paper as possible. All filter papers were burned to recover residual palladium; doing this is even more
important with platinum. Alternatively, since ammonium chloroplatinate is insoluble in ethanol, EtOH can be used to rinse of residual water then dry
the salt with vacuum.
Two methods of reduction were performed to show the advantages and disadvantages of each. Half of the palladium was reduced via pyrolysis of the
ammonium salt which takes advantage of thermal instability of the ammonium salt. This goes from palladate to palladite to palladium metal, all the
while producing ammonia and hydrochloric gases, which means copious quantities of ammonium chloride smoke and chlorine. It is a cheap, effective
method if done properly. If rushed, some of the salt can actually sublimate away, which means the loss of an expensive metal. This method of reduction
is less favorable for palladium than platinum because palladium is prone to oxidation at high temperatures, forming the green-black mix of Pd(II) and
(IV) oxides. Platinum does not oxidize at anywhere close to the temperatures used in this process, so it can be heated to a high temperature.
Essentially, one must heat the salt until it no longer off gases, and there will be formation of brown Pd sponge. After the pyrolysis is complete, the
Pd should be rinsed first with HCl, then with distilled water. Moisture is removed by heating until the sponge is free flowing or by rinsing with
absolute ethanol; yield 42.8139 g Pd. Shown below are pictures of the pyrolysis process.

Roughly half of all the dried palladium precipitate. Only the Pd was recovered with pyrolysis, the Pt was just reduced with AF system.

Much ammonium chloride and chlorine gas is evolved. Darkening is a good thing. Do not overheat or the fumes will no longer be white, but gray in
colour, indicating that your Pd is gas phase transporting out of the dish and out of your wallet. A cold beaker bottom is a good way to check if this
is happening, if white, it's only ammonium chloride (very good), if dark, it is Pd metal (very bad, whoops!)

At about 500*C the process is starting to finish. Soon all of the ammonium chloride will be gone, and a brown powder will be left. I did this on a
hotplate and used Kaowool ceramic fiber to keep the heat in, but a good Bunsen burner could be used.
The other method used for both platinum and half of the palladium is the ammonium formate reduction system where a slightly acidic ammonium formate
solution is heated to about 90*C and added to a similarly hot solution of ammonium palladate. Ammonium formate is easily made from formic acid and
ammonia, simply use pH paper or meter to determine how much formic acid to add (very exothermic process, do in a tall beaker). The precious metal salt
is dissolved in boiling water and added to a vigorously stirred 90*C/192F solution of acidified ammonium formate solution. Much effervescence occurs
as the formate is oxidized to CO and the palladium is instantly reduced to a fine, inky suspension of palladium black. This must be vacuum-filtered
with very slow, high quality filter paper, and rinsed with distilled water, and HCl several times. The Pd metal powder can then be dried with vacuum,
heat, or EtOH; yield 48.2757g. (Unfortunately, the photographs of the formate reduction off of my digital camera were missing. Same for some of the
other photos, darn!) The Pt chemistry is identical, except it takes a little longer for the reduction to occur (higher temperature would help). This
method is highly effective, but unfortunately gives very finely powdered precipitates, and is more expensive to use. I did not show the use of
hydrazine this time, but eventually I will demonstrate the use of N2H4 and its salts for PGM reductions.
Pd Chemistry—generalized reduction pathway, pyrolysis
(NH4)2PdCl6 -->(NH4)2PdCl4 + Cl2 --> Pd +Cl2 + 2NH4Cl
Ammonium formate route:
(NH4)2PdCl6 + NH4HCO2 --> Pd + NH4Cl +CO +H2O
Procedure-Platinum, Mo crucible, ink
The goal is to separate the Mo crucible from the stuck inside it. Pt is a very inert material, and is difficult to dissolve in hot aqua regia while in
cold aqua regia, it is barely affected by NOCl and can be considered insoluble. Molybdenum is not quite as inert, but it forms an inert, clingly,
bothersome oxide, MoO3 which can only be removed mechanically, or by dissolving in NH4OH. Nitric acid attacks molybdenum rather quickly, but this only
lasts for a few minutes because the surface becomes covered with a yellowish growth of oxide, preventing further attack. The crucible was added to
warm nitric, and continuously scraped to remove the oxide, but it was bothersome. The crucible was also removed periodically to soak in ammonia water
to remove oxide, but it was also found to be impractical. Therefore, the crucible was cleaned as well as possible, and then added to 60mL of 15*C aqua
regia which rapidly attacked the crucible, but did not faze the platinum. In about 4 hours, the molybdenum crucible turned into an orange-red crucible
shaped mass of mixed oxides, the solution was yellow-orange. Upon cutting into the oxide layer, it was found that a thin layer of metal remained, the
rest was a chalky oxide. Close to that metal the oxide layer was blue-violet in colour, indicative of molybdenum (IV) oxide. All of the oxide was
removed as best as possible with a spatula and wash bottle of distilled water, yielding a chunks of obvious platinum, and Mo-Pt bimetallic strip
(assumed to be the Pt liner and unreacted Mo). The Mo was determined by seeing a reaction when added to nitric acid, which also removed the last few
milligrams of Mo. The Pt was then filtered, rinsed, massed (2.8417g) and dissolved in hot aqua regia. The putty-like ink was rinsed with solvent,
dried, massed, then was broken up into small chunks and added to 60mL of aqua regia and boiled for approximately 14 hours until all finally dissolved.
This solution was combined with the solution obtained from the Mo crucible, boiled down to the acid chloride syrup, then precipitated with ammonium
chloride as a canary yellow ammonium hexachloroplatinate (IV), which was reduced using the ammonium formate process, yielding an inky black suspension
of Pt. That settled over night, was vacuum filtered using slow filter paper, rinsed with acids, then put in a dessicator and removed for massing and
bottling. The chemistry is identical.

The platinum 'ink' dissolving in aqua regia.

The molybdenum (VI) oxide removed from the crucible. Note the yellow colour of dilute Pt (+4).

The platinum extracted from the Mo crucible boiling away in very hot aqua regia. Cold aqua regia has virtually no effect on Pt unless it is a very
fine powder. Even these 1-3mm sized pieces took over 14 hours to dissolve completely.
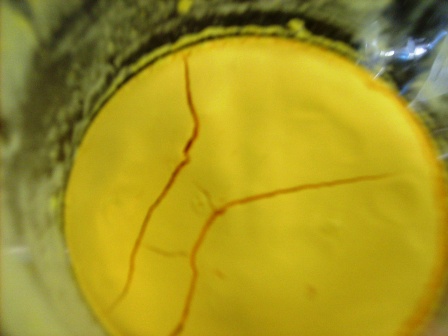
A picture of the precipitate, (NH4)2PtCl6, a beautiful canary yellow. Dry in dessicator, and then remove and mass.
Right here let me stop to show what a positive qualitative test for Pt looks like. I utilized a few crystals of stannous chloride dihydrate in
hydrochloric acid. I did this with my own Pt to exemplify how this test can be used to check for values in a solution that has been precipitated
already.

Before the test, acidified SnCl2 on the right, Pt ''suspect'' on left. If you do not acidify the tin (II) chloride, it will hydrolyze.

Positive test, note the colour.
Procedure-sputtering target
A MnSbPt sputtering target of unknown Pt composition was massed, then placed in a PTFE beaker, and 100mL of cold aqua regia was added. This produced
an immediate reaction, and the vessel needed to be cooled. An orange colour was noted, but quickly turned to a green-grey liquid the consistency of a
fluid mud. Upon adding more hydrochloric acid, the colour would become more orange, thus it is obvious that it was the antimony hydrolyzing to its
oxychloride and oxide. This would prove to be problematic because the antimony based precipitate was difficult to separate from the heavy gray
platinum on the bottom of the beaker. Repeated filtrations using concentrated, hot HCl removed most of the Sb contamination. The rest of the antimony
was sublimated off at 1000oC. It yielded only a couple grams of Pt metal, which was again dissolved in aqua regia and reduced using the above outlined
procedure. If the MnSbPt target was on an equimolar elemental basis, then the target would be roughly half platinum by mass (~34g), sadly however this
was not the case.

The sputtering target in a 250mL PTFE beaker. The aqua regia was kept cool to avoid any incidental Pt dissolution.

After adding aqua regia, quickly turns a nice dark red colour, then slowly changes to orange, then to yellow-green.

Note the dark gray Pt layer on the bottom of this glass beaker, also see the antimony compounds on top as a lighter powder. Turbid solution too, damn
antimony.
Final Yields
Molybdenum crucible and Pt ink yielded of 22.6153 g Pt
Platinum chunk with stuck-on graphite crucible base yielded 30.2420 g Pt
MnSbPt sputtering target: 66.3767g only~1.7 g Pt
All palladium materials yielded 101.1 g Pd
[Edited on 30-12-2007 by Fleaker]
Neither flask nor beaker.
"Kid, you don't even know just what you don't know. "
--The Dark Lord Sauron
|
|
|
guy
National Hazard
Posts: 982
Registered: 14-4-2004
Location: California, USA
Member Is Offline
Mood: Catalytic!
|
|
beautiful
|
|
|
noxx
Harmless
Posts: 25
Registered: 27-12-2007
Location: Québec, Canada
Member Is Offline
Mood: Raffinate
|
|
You like platinum group metals right ? 
Ha... I hope I would get all that lab gear at home....
Good post by the way !
Knowledge is the Key.
|
|
|
guy
National Hazard
Posts: 982
Registered: 14-4-2004
Location: California, USA
Member Is Offline
Mood: Catalytic!
|
|
I hope you actually do something interesting with their chemistry
|
|
|
DerAlte
National Hazard
Posts: 779
Registered: 14-5-2007
Location: Erehwon
Member Is Offline
Mood: Disgusted
|
|
Nice chemistry, Fleaker, well presented. You have a pretty good lab! Looking forward to your efforts with Os,Ir,Re, Ru. That Pt is worth a fair bit!
And the Pd too, not sure of its price these days.
Regards,
Der Alte
|
|
|
Fleaker
International Hazard
Posts: 1252
Registered: 19-6-2005
Member Is Offline
Mood: nucleophilic
|
|
Thank you all. I plan on covering how to put them in solution, selectively precipitate, and then reduce. I plan on covering the use of hydrazine and
its salts, borohydride, formate, cementation with other metals, and for Os, Ir, Ru, Re the use of hydrogen and high temperatures for reduction.
Eventually I hope to cover the preparation of various salts and complexes and look at the coordination chemistry. I do not know how soon that will be,
or if it will be.
Neither flask nor beaker.
"Kid, you don't even know just what you don't know. "
--The Dark Lord Sauron
|
|
|
12AX7
Post Harlot
Posts: 4803
Registered: 8-3-2005
Location: oscillating
Member Is Offline
Mood: informative
|
|
Awesome.
I, too, have a golden colored solution, water currently worth about $300/gallon -- PtCl6(2-) solution. I'm ready to try platinizing a titanium anode.

Tim
|
|
|
Waffles
Hazard to Others
Posts: 196
Registered: 1-10-2006
Member Is Offline
Mood: No Mood
|
|
Its pretty rad to see all the work put into my metals.
Fleaker knows his stuff, eh?
\"…\'tis man\'s perdition to be safe, when for the truth he ought to die.\"
|
|
|
noxx
Harmless
Posts: 25
Registered: 27-12-2007
Location: Québec, Canada
Member Is Offline
Mood: Raffinate
|
|
Ya he does lol
Knowledge is the Key.
|
|
|
Intergalactic_Captain
Hazard to Others
Posts: 227
Registered: 4-9-2004
Location: somewhere where i don\'t know where i am
Member Is Offline
Mood: frabjous
|
|
Damn...Nice work there. What's that, something like $3000 USD spot value for everything? The best I've done so far is about an ounce of silver
recovered from miscellaneous coin and jewelery scrap.
If you see me running, try to keep up.
|
|
|
12AX7
Post Harlot
Posts: 4803
Registered: 8-3-2005
Location: oscillating
Member Is Offline
Mood: informative
|
|
| Quote: | Originally posted by 12AX7
Awesome.
I, too, have a golden colored solution, water currently worth about $300/gallon -- PtCl6(2-) solution. I'm ready to try platinizing a titanium anode.
Tim |
Speaking of platinum, I am currently digesting the residues (black and metallic flakes) from my less successful plating attempts.
Right now, I have a hot solution of HCl with some KNO3 added. Doesn't seem to be getting much redder. I need a heating mantle...
Edit: I think I'm mostly done with it, the boiling that is. I added a pinch of KNO3 towards the end, which resulted in an immediate release of
unknown gas (it didn't appear to be colored, e.g., NO2, Cl2) and an insoluble green material precipitated, rather than the yellow K2PtCl6 I was
expecting (and had washed from the solids earlier).
I don't know what this compound is, any ideas? Pt oxide is clearly absurd in acid, perhaps nitrito somehow?
Tim
[Edited on 1-16-2008 by 12AX7]
|
|
|
jokull
National Hazard
Posts: 506
Registered: 22-2-2006
Location: Everywhere
Member Is Offline
Mood: Ice glassed
|
|
Hi.
This thread is quite interesting, although I'm not experienced with recovery. Searching over the internet I found this book:
https://sciencemadness.org/talk/post.php?action=reply&fi...
I hope someone find it useful as I do.
|
|
|
noxx
Harmless
Posts: 25
Registered: 27-12-2007
Location: Québec, Canada
Member Is Offline
Mood: Raffinate
|
|
Jokull, I don't have access, I think I must be a special member of something...
Anyways, visit the goldrefiningforum if you want to learn more about PMs recovery/refining.
www.goldrefiningforum.com
Knowledge is the Key.
|
|
|
|













