
Quote: Originally posted by Mailinmypocket  |
| Quote: Originally posted by blogfast25 |
 .
That should really rule out any temperature problems.
.
That should really rule out any temperature problems.| Quote: Originally posted by elementcollector1 |
| Quote: Originally posted by blogfast25 |
| Quote: Originally posted by Antiswat |
| Quote: Originally posted by blogfast25 |
| Quote: Originally posted by Mailinmypocket |
| Quote: Originally posted by elementcollector1 |
| Quote: Originally posted by blogfast25 |
| Quote: Originally posted by elementcollector1 |

| Quote: Originally posted by elementcollector1 |



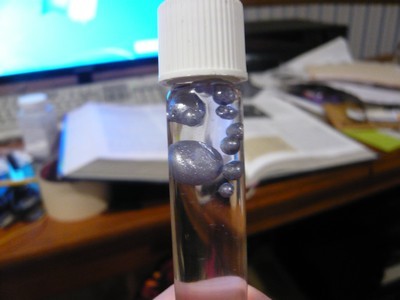

 very nice though, good job! Ill have to read over your last couple posts in
detail when I have time, but this is very promising!
very nice though, good job! Ill have to read over your last couple posts in
detail when I have time, but this is very promising! 
| Quote: Originally posted by m1tanker78 |
| Quote: Originally posted by elementcollector1 |
| Quote: Originally posted by elementcollector1 |
| Quote: Originally posted by Lambda-Eyde |
| Quote: Originally posted by blogfast25 |
| Quote: Originally posted by blogfast25 |
| Quote: Originally posted by blogfast25 |
| Quote: Originally posted by Lambda-Eyde |
| Quote: Originally posted by blogfast25 |
There's lots more great information here, and the document is really
no substitute for reading the thread in its entirety.| Quote: Originally posted by MrHomeScientist |
| Quote: Originally posted by MrHomeScientist |
| Quote: Originally posted by MrHomeScientist |
| Quote: Originally posted by MrHomeScientist |
| Quote: Originally posted by elementcollector1 |
| Quote: Originally posted by elementcollector1 |

| Quote: Originally posted by MrHomeScientist |