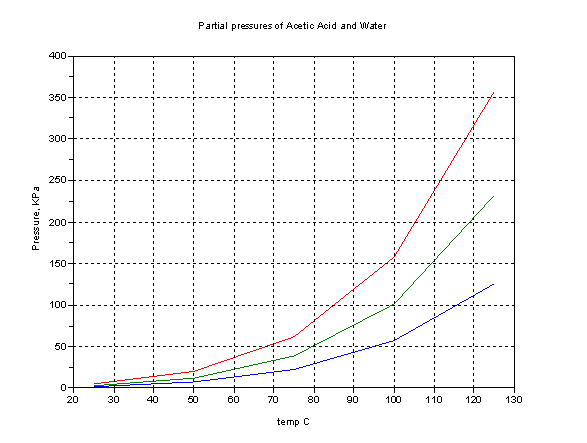
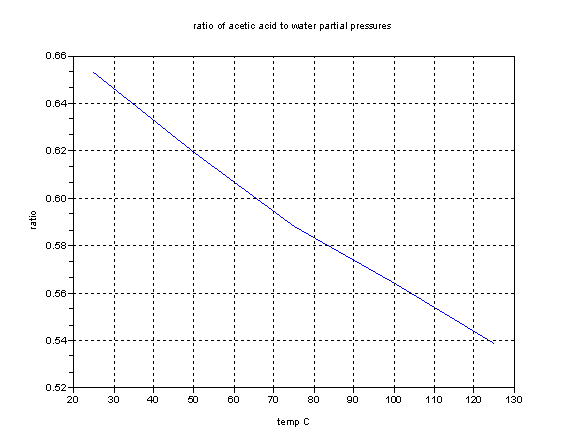
I entered the single word "lye" in Google and got 493,000 results. The relevant results were very much in favor of "lye" meaning
KOH solution, not NaOH, most especially that made the old-fashioned way, by lixivating wood ash, which contains at least 10 times more K than Na,
|