manufacture of cyanide by Erlenmeyer's process of
fusing with metallic sodium, when the following changes take place :
K4Fe(CN)6 + 2Na = 4KCN + 2NaCN + Fe.
In this process, first worked between 1890-1900, all the cyanogen is recovered in the form of sodium or potassium cyanide, the sodium cyanide being
technically of the same vahie as the potassium cyanide, provided the CN content is the same.
The process is carried out as follows:
In covered iron crucibles, some 30-40 cm. in height dehydrated potassium ferrocyanide, K4Fe(CN)6, is mixed with the proper amount of metallic sodium
in the form of short bars, and the crucible is then heated over a free fire until the contents are completely fused. The molten contents of a number
of these crucibles are next poured into an
iron crucible, heated by direct fire as before, but provided with a filtering arrangement made of spongy iron (obtained in the above-mentioned melting
process), below which are outlet tubes. The cyanide is forced_through this filter by means of compressed air and a compressing piston, as it flows
away from the filtering crucible solidifies to a white crystalline mass. It contains some cyanate, KCNO or NaCNO, along with a little alkali
carbonate. Nevertheless, in practice the cyanide is always valued on the basis of the parts of NaCN are technically equivalent to ioo g. KCN, the
cyanide can be placed on the market as " 100 per cent. KCN" in spite of the presence of these impurities. It is only the CN which counts, technically
; whether the CN is united with K or Na... |






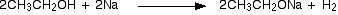
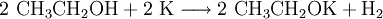

 .
.