Fries rearrangement. Leonte, Mircea; Beschia, Magda; Pascaru, Elvira; Stoica, Maria. Polytech. Inst., Galati, Rom. Studia Universitatis
Babes-Bolyai, Chemia (1963), 8(1), 291-6. CODEN: SUBCAB ISSN: 1224-7154. Journal language unavailable. CAN 61:68907 AN 1964:468907
CAPLUS
Abstract:
Starting from the hypothesis that the strength of the acid catalysts plays an essential role in the initiation of the Fries rearrangement, the
capacity of 70% HClO4 to catalyze the reaction of phenol acylation was investigated, since the only case of the use of this acid was the unsuccessful
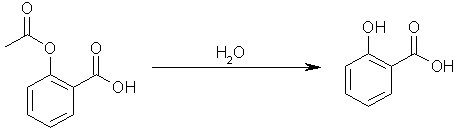
attempt of Dorofeenko (CA 55, 24518i) to acylate phenol and resorcinol, which gave the esters instead of the ketones. Phenol (50 g.) was treated with
45 cc. Ac2O and 10 cc. 70% HClO4, the mixt. heated 30 min. on a water bath, left 24 h. at room temp., neutralized with 20% Na2CO3, 20 g. NaOH in 100
cc. H2O added, the aq. alk. soln. extd. with ether, then neutralized with 20% HCl, reextd. with ether, the ether evapd., and the product distd. to
give 51% 2-hydroxyacetophenone, b7 86, and the distn. residue dissolved in 20% NaOH, boiled 30 min., cooled, pptd. with 10% HCl, and recrystd. from
H2O with animal charcoal, to give 20.5% 4-hydroxyacetophenone, m. 108-9. Parallel Fries rearrangement of PhOAc under the same conditions gave the o-
and p-isomers in 38 and 19% yields, resp., thus confirming that the ester was formed at first and was then rearranged to the corresponding ketone.
Similar treatment of o-cresol (3 h. at 120) gave 55.5% 2-hydroxy-3-methylacetophenone, b10 105, 31% 3-methyl-4-hydroxyacetophenone. Treatment of
m-cresol (1 h. with no external heating, then 48 h. at room temp.) gave the o- and p-isomers, 62.8% 2-hydroxy-4-methyl- and 6%
4-hydroxy-2-methylacetophenone, b7 103, and m. 128% resp. Similar treatment of p-cresol (heating 1 h. at 100 then keeping 48 h. at room temp.) gave
53% 2-hydroxy-5-methylacetophenone, b7 101-3. Treatment of resorcinol (cooling, strong agitation, then 24 h. at room temp.) gave after neutralization
with Na2CO3 90% 2,4-dihydroxyacetophenone; similar treatment of resorcinol monoacetate gave 83% same product. Treatment of pyrocatechol (refluxing 3
h. on a water bath) gave 58% light-violet 2,3-dihydroxyacetophenone, m. 97.
Identical treatment of pyrogallol gave 44% 2,3,4-trihydroxyacetophenone, m. 173. Treatment of .alpha.-naphthol (1 h. at 115-20 on a sand bath) gave
after steam-distn. part of the 1-hydroxy-2-acetylnaphthalene formed, m. 100; further boiling of the resinous residue 1 h. in NaOH, neutralization, and
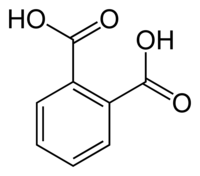
steam-distn. gave some further product, for a total yield of 40%. Salicylic acid was first dissolved in Ac2O-HClO4 at 100, the mixt. kept 0.5
h. at 120-30, cooled, H2O added, and the ppt. filtered off and recrystd. (H2O) to give 20-5% 3-carboxy-4-hydroxyacetophenone, m. 209-10,
phenylhydrazone m. 260; semicarbazone m. 222. Heating the reaction mixt. 1.5-2 h. at 120-30 and similar purifn. gave 71%
2-hydroxy-3-carboxyacetophenone, m. 148-9; phenylhydrazone m. 218; semicarbazone m. 240. The cause for the limited usefulness of HClO4 as a
catalyst in the Fries rearrangement was hence attributed to the oxidizing nature of this acid, which appeared only >170 for long durations in the
case of phenol acylation, but did not appear when the reaction temp. was kept <140 in the case of the Fries rearrangement. Its advantages over
other acid catalysts were: low amt. needed (0.1-0.2 mol per mol phenol compared to 1-2 mol AlCl3); no solvent needed as is the case with AlCl3 (the
use of solvents in the case of HClO4 did not improve the yields); and the preferential catalysis to the o-isomer, which in the case of some
phenols-pyrocatechol, pyrogallol, and salicylic acid-was the only isomer obtained, in contrast to the p-isomer obtained when AlCl3 was used, while in
the other cases the yields of o-isomer were greater than and the yields of p-isomer smaller than when AlCl3 was used as catalyst.
|



 ) would actually form a skin and form 'crevices' through it's self, like an over
baked milky pudding. At hydrolysis, absolutely nothing would happen. No fuming, no breakdown of the cake. I had to warm the solution to just above
room temperature. Again, no evolution of gas. But the cake would then breakdown into fine grains of aluminum.
) would actually form a skin and form 'crevices' through it's self, like an over
baked milky pudding. At hydrolysis, absolutely nothing would happen. No fuming, no breakdown of the cake. I had to warm the solution to just above
room temperature. Again, no evolution of gas. But the cake would then breakdown into fine grains of aluminum.
 --->
--->