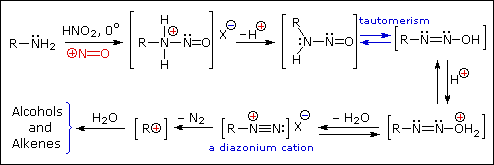
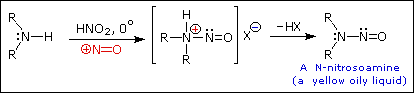The person placed 1 mol of GABA (103g), 69g of NaNO2 (food grade), and 230ml DH2O to a 1L round bottom flask. They then added 115ml HCL (technical
grade) dropwise with a rate of ~1 drop/8 seconds with heavy stirring. Once the HCL was fully added stirring continued for ~1hr, and was then
transferred to another flask. This procedure was then repeated in order to obtain ~900ml of rxn mixture. The solution was allowed to sit overnight
with light stirring to allow the rxn to come to completion (room temperature was ~0 Celsius due to weather). After ~20hr the rxn mixture was placed in
a room temperature environment. After the 24hr period the rxn mixture was placed into a 1L rb flask again, and was set up for simple distillation.
The distillate collected was ~400ml of crystal clear liquid. It had a pH of ~6-7, and no noticeable odor. The distillate was brought up to a pH of
~7-8 using NaOH (food grade). This fluid was then transferred to a beaker and boiled down to 100ml and allowed to cool. No solidification occurred so
it was further boiled down to 50ml, and cooled. No solidification - the liquid now had a slight yellow hue to it, and a slightly salty taste, and ever
so slightly more viscous.
The post rxn mixture was placed in the freezer to precipitate any NaCl, and then decanted off. It was then placed back into a simple distillation
until bumping occurred. Then placed back into the freezer to encourage more NaCl precipitation. Once decanted for the final time ~350ml of post rxn
fluid was placed into a 500ml sep funnel and washed with EtOAc in 200ml portions 5x times.
The EtOAc was placed into a 1L rb flask with a fractional distillation column. Once all of the EtOAc was collected from distillation ~25ml of dark
brown liquid remained. The 25ml was then placed into a micro distillation setup and distilled under vacuum ~5ml of crystal clear liquid was recovered
with a pH of ~5.
The discard fluid from the EtOAc washes was now comprised of over 50% salt, and the remaining liquid was a few shades off black.
|