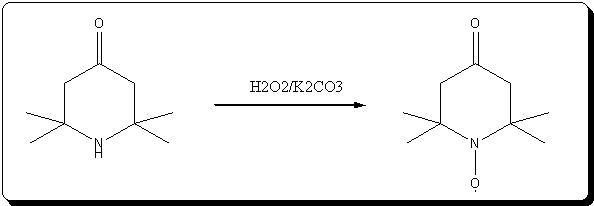
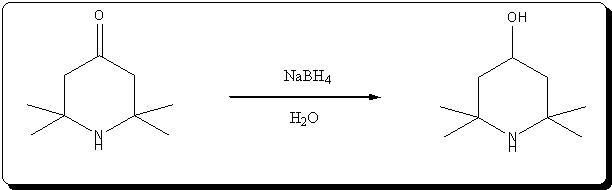
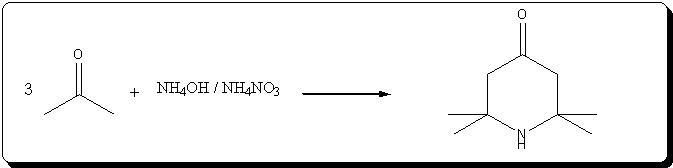
A. 2,2,6,6-Tetraperdeuteromethyl-4-oxo-3,5tetradeuteropiperidine (Triacetoneamine-d16) (V)
A mixture of ammonium-d4 chloride (3.45 g, 0.06 mole), acetone-d6 (99.5 atom %D, 12.5 ml, 0.15 mole), anhydrous sodium carbonate (3.18 g, 0.03 mole)
and magnesium oxide (3.0 g) was added to a 250 ml round-bottomed flask. The flask was capped with a rubber septum and wired, then the reaction mixture
heated in an oil-bath at 50° C. for 3 days. After cooling, 20 ml of acetone was added to the reaction mixture and the resulting mixture was filtered.
The recovered solid was crushed into powder, washed with 15 ml of acetone and then filtered with suction filtration. The combined filtrates were
concentrated to dryness. The resulting red liquid (7.2516 g) was distilled under reduced pressure to obtain 4.7480 g (56.7%) of a bright yellow liquid
(b.p. (boiling point) 54°-55° C./1.9 mm Hg) that solidified when chilled in a dry ice/acetone bath. The solid product subsequently was used without
further purification. Recrystallization of an analytical sample from anhydrous diethyl ether yielded white crystals, mp. 57°-58° C. [lit. 58° C.];
IR(KBr, cm-1):3580(m), 3260(m), 2220(m), 1700(s), 1530(w), 1265(s), 1140(m), 1050(m), 930(w); 13 C-NMR (CDCl3): 31.03(m), 53.50(m), 54.88(s), 211.19
(s).
|



 I since procurred the stopper from my local hardware store!
I since procurred the stopper from my local hardware store! and I
added the 100 ml acetone needed.
and I
added the 100 ml acetone needed.
































 :
: