Ways of producing H2SO4 have been discussed many times here on SMDB, but there is still no simple way to produce it.
You have a few choices when trying to get your hands on H2SO4:
1. Buy Super Impure Sulfuric Acid Drain Opener
2. Build the Sulfur Burner proposed by Axehande
3. Electrolyse a Sulfate Salt as discussed by seraniyde.
4. Boiling down battery acid. (DANGER!)FUMES + CORROSIVENESS
There has got to be a better way!
Can this be another possibility...
Sodium Bisulfate seems like an undiscussed route to H2SO4. IT can be found as pH Minus and made from drain opener H2SO4 +NaOH. When added to HCl it
gives off SO2, but that doesn't held much either since you need Vanadium Pentoxide to convert to SO3.
alternatively...WIkipedia says "Sodium bisulfate behaves, to some degree, as if it were a complex of sodium sulfate with sulfuric acid. This is
evident if either the anhydrous form or the monohydrate come in contact with ethanol, which causes them to separate into those two components."
So Sodium Bisulfate with Ethanol will cause a separation into sodium sulfate and H2SO4! BINGO? Sulfuric acid right there no?
The only think I see wrong is that wouldn't that make Ether with the H2SO4 then? If another Alcohol was used would this not happen.
Lastly, check this out...
2NaHSO4 → Na2S2O7 + H2O @ 315°C
Na2S2O7 → Na2SO4 + SO3 @ 460°C
460 Celsius is very achievable.
What do you guys think about the three methods proposed here? I think method two is easiest, but there is the Ether problem.
thanks
MericadBromicAcid - 24-10-2006 at 14:55
The sodium bisulfate / ethanol route has been discussed here before, although I don't know if a final consensus on it was reached. It has been
attempted here. Sulfuric acid really is difficult to make, thank god it is an industrial chemical otherwise most of us would never see it.not_important - 24-10-2006 at 20:40
Quote:
Originally posted by mericad193724
Ways of producing H2SO4 have been discussed many times here on SMDB, but there is still no simple way to produce it.
You have a few choices when trying to get your hands on H2SO4:
1. Buy Super Impure Sulfuric Acid Drain Opener
(snip)
4. Boiling down battery acid. (DANGER!)FUMES + CORROSIVENESS
That is what distillation apparatus is for, don't go around boiling acids in open pots in the kitchen.
Quote:
(snip)
So Sodium Bisulfate with Ethanol will cause a separation into sodium sulfate and H2SO4! BINGO? Sulfuric acid right there no?
The only think I see wrong is that wouldn't that make Ether with the H2SO4 then? If another Alcohol was used would this not happen.
Sorry, it will happen with most alcohols, or the alcohol will dehydrate to a corresponding alkene. I've made di-isopropl ether from isopropanol (kids,
don't do this at home, it forms peroxides as you watch).
I may have to try this. I think the way to go would be to chill everything, filter off the Na2SO4 after allowing the reaction to occur, add a measured
ice water and removed the alcohol under reduced pressure. You'll still end up with somewhat dilute H2SO4, and still need distillation gear. Or can
ethanol be distilled from strong H2SO4 at low pressures without problems?
Quote:
Originally posted by BromicAcid
Sulfuric acid really is difficult to make, thank god it is an industrial chemical otherwise most of us would never see it.
This is too true. Even the old 19th century chemistry books say that making H2SO4 is an industrial process.
One more alternative, although not OTC, would be to use selective ion membranes in a form of electrodialysis. For example, using a solution of sodium
sulfate you would generate sulfuric acid and sodium hydroxide. Both would be contain some Na2SO4, and would not be very concentrated. Running each
solution separtly through the electrodialysis would remove all but the smallest amount of Na2SO4, after which conventional concentration methods would
be needed to get solutions strong than around 30%. This would also work with NaCl to make NaOH and HCl, and ammonium sulfate to make aqueous ammonia
and sulfuric acid.
I've never tried to track down the membranes, so I've no idea how expensive they are; although I know that small units for laboratory purposes are
made. It takes more energy, and expensive electrical energy at that, to make the products that the conventional industrial processes, so it is not
used for that purpose except in special cases.Tacho - 25-10-2006 at 03:47
Boil down battery acid.
I assume you want reasonably concentrated acid. All methods of production "from scracht" will either give you dilute acid that has to be boiled down
anyway or be far more dangerous (*) to work with than boiling down battery acid.
I have boiled down battery acid a few times and the distilate was barely acidic at all, meaning that all the acid remains in the boiling solution. I
think the slight acidity in the distilate was due to the mist formed by boiling, some of which reached the condensor. The only concern here is that
you have hot concentrated acid in the boiling flask, if it breaks or spills...
Sometime ago I posted a concentration table for those boiling down battery acid. I even attached a spreadsheet. It's here:
(*) Axehandle's (brilliant anyway) method involves the production of SO3 at about 450ºC at some point. If boiling-hot concentrated sulfuric acid has
been called "liquid chainsaw", SO3 at 450ºC could be called "gaseous chainsaw".BeerChloride - 25-10-2006 at 07:00
Mericad, there's one more - freezing. Has anyone tried this? I've been meaning to try dry ice which I can get for $1 per pound. I think sulfuric acid
freezes very slowly, though. It also may take a couple of cycles of removing crystals, melting, and re-freezing. The monohydrate (about 86%) has a mp
of near 8 C, and anhydrous is near 10 C, with a pretty flat transition between. It's very easy to boil down to the monohydrate (about 105 C), VERY
hard to remove water beyond that by heat. So that could make for a simple scheme. In principle, it could be done in a regular freezer, but I put about
5 ml of the brown stuff in a -20 degree freezer for 8 hours and nothing..unionised - 25-10-2006 at 10:20
So far as I'm aware threre is not "society for the prevention of cruelty to alcohols" so if they get trashed to ether and or ethene I don't care.
Ethanol is cheap enough.
"460 Celsius is very achievable."
That's probably a moot point, but if you are happy enough with SO3 at 460C then distilling H2SO4 should be a walk in the park.
Dilute sulphuric acid isn't that hard to make; SO2 (from burning sulphur) and aqueous H2O2 would work nicely. SO2 and ozonised air or ordinary air
over a Pt catalyst works fine, but trapping the O3 is a problem.
I guess the easy way is to buy battery acid and boil out the water or distil drain cleaner (I don't think that idea's very apealing unless your "lab"
is separate from your house)mericad193724 - 25-10-2006 at 11:44
I think I will try to boil down Battery Acid. I just bought 900ml for $4 US, its around 40% so that is a good and cheap start. I know boiling it is
pretty nasty because it bumps a lot. I have a borosilicate distillation setup (Graham Condenser, 500ml RBK).
Not_Important: I got real glassware, boiling H2SO4 in the kitchen in a stainless POT is a painful suicide!
Freezing sounds too easy. I don't have dry ice, where can you get it?
Will hot conc. acid frost up my glassware? Will a Cooking Oil bath suffice or do I need Sand?
Some of you have done this (beerchloride mentioned it once), any tips?
MericadTacho - 25-10-2006 at 14:46
It will not damage your glassware.
Use boiling stones!evil_lurker - 25-10-2006 at 16:25
They'll hook ya up with 98% ACS grade sulfuric for $13.80 per liter if you buy a 4 pack.
And the best part is they ship it ORM-D so no Hazmat Fees!mericad193724 - 25-10-2006 at 16:43
The only boiling stones I got are CaCO3 chips and those obviously won't go well with H2SO4. I heard broken glass works too (break some glass bottles).
What is special about a sand bath? Can you use any other try of powder material that doesn't decomp such as Plaster of Paris or Na2CO3?
I will do distillation of Battery acid tomorrow if I can get this cleared up. (With Pics!)
Mericadnot_important - 25-10-2006 at 17:34
Even strong H2SO4 will not damage borsilicate glassware.
Sand because it is inert and it is not a powder. Powders tend to cling to the glassware, and often are worse heat conductors than sand (which isn't
that great). Sand is also much less expensive than those alternatives you named.
Inert, unlike plaster of paris which absorbs water from the air, or sodium carbonate that reacts with acids.
Table salt might work, but compare its thermal resistance to SiO2.
Calcium carbonate is generally a lousy boiling stone. Reacts with acids, and is basic so it can futz up some organics. Broken glass is OK, but not as
good as high surface area boiling stones. Not that we can affort it, but once platinum wire was recommended as an anti-bumping device when heating or
distilling sulfuric acid.
Battery acid is reasonably strong, some spa & pool "ph Down" is 10% sulfuric acid.
The boiling point of 80% sulfuric acid is close to 200 C, 40% close to 105 C
Edited to add: While you may have proper glassware, I've read postings elsewhere of people that were going to did try to use glass/ceramic pots on
the stove for boiling down battery acid.
[Edited on 26-10-2006 by not_important]leu - 25-10-2006 at 18:26
Quote:
The only boiling stones I got are CaCO3 chips and those obviously won't go well with H2SO4. I heard broken glass works too (break some glass bottles).
Method for Making Boiling Stones
Clean and efficient boiling stones are easily prepared by melting pieces of glass wool held with forceps in the flame of a Bunsen burner until a
sintered chip with a smooth surface is formed.
[Edited on 26-10-2006 by leu]Magpie - 25-10-2006 at 18:40
For good boiling stones the easy and cheap way:
Get an unglazed clay flower pot and break it into small pieces (~ 1/8" dia).not_important - 25-10-2006 at 23:43
WhotDaL...
I did some searching, and didn't find this method, so let us toss Yet Another Route to Sulfuric Acid out.
There's a proposed method of generating hydrogen thermally. H2SO4 is heated about 800 C to produce O2, H2O, and SO2. This is then mixed with iodine
and H2O at ~120 C, the resulting Bunsen reaction leaves the O2 alone and produces HI and H2SO4. The H2SO4 is recycled back into the decomposition
stage. The HI is heated to ~ 350 C to break it down into H2 and I2, with the I2 going back to the Bunsen stage.
So what if we replace the H2SO4 decomposition with a sulphur burner, and the HI decomposition with an air oxidation of HI tp H2O + I2 ?
I must confess I haven't read enough on the Bunsen reaction to see what conditions are eneded to get good yields and allow easy separation of the HI
and H2SO4. Perhaps this could be make into a dual catalyst system I2 serving to transform SO2 and H2O into H2SO4, and (something) + O2 converting HI
into I2 and H2O.
maybe a dumb idea, I need to look at the reaction specifics in more detail.
[Edited on 26-10-2006 by not_important]Tacho - 26-10-2006 at 04:37
About boiling stones:
I never cease to be surprised by some good members of this board mentioning broken glass or other nonporous materials as boiling stones. Some mention
the sharp edges as beeing good nucleating points.
In my experience, the result using porous ceramic are so much better than nonpourous materials that, comparing the results, is hard to believe that
any nonporous material is working at all. Porous materials give a flow of bubbles like those aquarium stones.
My oppinion is that boiling stones have to be porous enough to trap some air or keep some (gaseous) vapour inside.
This opinion is backed up by two facts I noticed while doing experiments, both also mentioned in books like Vogel's or Zubrick's:
1- Boiling stones are no good in vacuum distillations. In this case you must use a capilar air inlet. If it was a matter of sharp edges or rough
surfaces, why would it be like that?
2- Boiling stones decrease activity sharply or completely if the boiling solution is cooled and then brought to boil again. You must add new boiling
stones. Again, this does not make sense under the sharp-edges-as-nucleating-points theory.
Also, IIRC, Vogel (third ed.) mentions the use of an iverted closed tube to act as a boiling aid. Like a small test tube (full of air) inserted upside
down in the boiling solution. I never tested this, but is coherent with my oppinion.
My theory about fact 2 is: in the begining of boiling, the porous(air filled) stone gives a liquid/gas inteface where the vapours can expand and
create bubbles. Eventually the air is substituted by the liquid's vapour which still provides an interface for the bubbling. But after cooled, the
vapour condenses and the liquid invades all pores, renderig the boiling stones useless.
I also tried teflon chips, but they floated. Good boiling stones must create their bubbles at the bottom, not only because there is where the hot
spots are, but also to create currents that prevent their formation.not_important - 26-10-2006 at 07:12
While a lot of places go along with the idea that it is the trapped gas that is the secret, t the same lab procedure books that say 'it's the gas'
also suggest such devices as sharp platinum bits or powdered glass fused to the inside of the flask, and have references to studies on such 'sharp
point' methods. Go figure.
Some of the sharp edge methods may actually be effective due to gas pockets trapped by the rough surface; the one that had the broken glass be made by
dropping red hot glass scraps into cold water and then oven drying them would certainly seem to lead to a lot of gas filled microcracks in the glass
fragments.
Cheap earthenware pottery may have problems when used with strong acids, although working well with organics. Unglazed porcelain, or at least mostly
unglazed, is better.Rosco Bodine - 26-10-2006 at 13:19
There are at least two main ways that boiling stones can work , porosity providing for adsorbed gases or a solvent which can act as a vapor source ,
being driven to evolution at somewhat below the boiling point of the surrounding mixture ......combined with mechanical movement of the stones against
each other caused by
the effervescence , the motion of sharp contact points
of the hard boiling stones causing miniature hot spots
by frictional contact , which results in a microenvironment
where boiling point is exceeded . The motion of the stones as they begin to rattle against each other in
the boiling liquid then further enhances the effect .....
and it does occur in a vacuum also . Fairly coarse granules of bone charcoal which have been presoaked
in toluene or benzene can work well as boiling stones
for vacuum distillations of organic materials . Broken
pieces of terra cotta flower pot , treated the same way
and mixed with the charcoal , and even some fairly large
pieces of broken glass , can together or separately be
effective , at slightly different induction points where
the effect is purely mechanical as for the glass , or
a combination of offgassing and mechanical as for
the porous materials . And then also there is the
reactive sort of boiling stone which may be metal or mineral and is partly consumed in the process , by
reaction with the component being boiled .....if some
sacrificial loss is acceptable for taming the boiling of the
mixture . A darker color and more heat absorptive property
for the boiling stone is also beneficial , as it is desired for the boiling stone to get hotter more quickly than the liquid in which it rests , so
that it acts as a local heat source in contact
with the liquid to be boiled .
[Edited on 26-10-2006 by Rosco Bodine]mericad193724 - 26-10-2006 at 13:43
I just tried my fist attempt at boiling battery acid to concentrate it....I didn't even get it to boil before I had to abort.
I was using a small kitchen pot that has that non-stick coating on the inside and some type of paint on the outside filled with fine sand. On this I
put my 500ml RBF. For boiling stones I broke a bottle and took about 5 1/4in pieces and scratched them up with 220 sand paper. These should provide
plenty of nucleation points. The heat source is a 1000W hotplate that gets RED hot. After about 5 minutes of heating there was copious amount of light
grey smoke coming from the sand or inside of the sand pot, can't tell which one. It smelled terrible so I stopped heating and called it a day.
Do you think it is the non-stick coating decomp. since the sand if not very good heat conductor so the pot got hotter than it is supposed to?
I will try again tommorow if I can firgure out the problem.
Mericadchromium - 26-10-2006 at 14:10
Your sand was probably contaminated with some organic matter.
If your hotplate gets red hot then you probably do not need sand bath at all. Use air bath instead - just fix your RBF half inch above hotplate.
For boiling stones i use pieces of broken kitchenware. In my experience glass, even if heavily scrached is not good enough. White clay? (i do not know
how it is actually called in english) of which common mugs and soup plates are made is very good if pieces have enough porous area without that common
enamel coating.
You also need very good ventilation. In my opinion - fume hood or do it outside. SO2 is not fun at all.
[Edited on 26-10-2006 by chromium]not_important - 26-10-2006 at 14:47
All three - teflon type coatings start to outgas at about 220 C, mostly short chain polymers, at around 300 C - distilling temperature for H2SO4 - the
fluorocarbon starts to break down into an interesting and fairly toxic mix.
The colourful outside coating on cookware can outgas at 400 C and above. Used cookware often has a coating of baked on and oxidised fats that smoke
when heated above 300 C
Unwashed sand usually has organics in it, even washed sand has a little.
So, for sand or air baths use uncoated, unpainted containers or first heat them to 500 C with very good ventalation. Don't use non-stick pans unless
you first mechanically remove as much of the non-stick coating as you can by sanding - perferablly all of it; then do the bakeout. Before using sand
for the first time, wash it with water several times, allow it to dry, then heat it to 500 C or higher; using cast iron pans to hold it and a self
cleaning oven to heat them may work, some self cleaning ovens depend on a catalytic coating and don't get as hot as the earlier models.
Don't boil sulfuric acid around anything metallic that you want to keep unless you have a really good draft pulling the fumes away.
They'll hook ya up with 98% ACS grade sulfuric for $13.80 per liter if you buy a 4 pack.
And the best part is they ship it ORM-D so no Hazmat Fees!
All VERY tentative but:
I might be able to beat that price if I get some in bulk. I know of a source for 18M H2SO4, GAA, 12M HCl, 16M HNO3, anhydrous hydrazine, both 85%
formic and ortho phosphoric acid, along with many dry chemicals (i.e. oxalic acid). DMF, DCM, acetone, Et2O also are available. Problem is, there's a
large deposit on the containers (several hundred dollars per stainless keg of 15 gallons capacity!). Also, ordering bottles to contain specific
things would be a problem. Some can go in HDPE, but some need glass (i.e. HNO3).
Here's my question though: is there enough of a market for this to justify spending several thousand dollars?
Assuming the above doesn't materialize, I've been meaning to get around to this oleum project. I plan to use a sulfur burner that I've designed myself
using a reservoir of liquid sulfur resistance heated to 120*C with an wick made of a high surface area, inert (mullite actually, some of you may know
it as kaowool) material. Pre-dried air (I have a washing bottle I can set up with conc. H2SO4) will be blown in at one point oxidizing the sulfur to
SO2. That will then be brought into the reaction chamber via the venturi effect*(V2O5 on more of that kaowool) which will be hopefully 2 inches in
diameter of stainless 316 flanged swagelock. That will be insulated and heated to sufficient degree to initiate the self-sustaining oxidation. The SO3
will then be lead into concentrated sulfuric acid reacting to produce oleum, H2S2O7.**
I have hopefully attached a crude pictorial explanation of the intended process.
I have it labeled also, here are what the letters correspond to:
a. heating vessel w/ thermocouple to control temperature
b. liquid sulfur
c. the kaowool ''wick''
d. the dry air inlets from drying column
e. Stainless 316 tube 3 ft/~1m in length by 2 inches/~5cm diameter
f. Insulation to facilitate heating of reaction tube
g. kaowool substrate which has had V2O5 deposited onto it
h. half inch/1.25 cm stainless piping that leads the SO3 into the a bubbler
i. sulfuric acid of approximately 16M concentration
j. cooling vessel filled with ice water
Open to and appreciating any criticism, ideas, and suggestions!
*The air required will be supplied by sending compressed air through sulfuric acid washing bottle. Flow rate to be determined.
**Exotherm of SO3 with water is too great and requires excessive cooling.
[Edited on 26-10-2006 by Fleaker]
Magpie - 26-10-2006 at 15:51
I had not played with molten sulfur for a long time until yesterday when I made some H2S in my hood for making aqueous Na2S. I used
to love to burn sulfur as a teenager in my parents basement. Fortunately I always seemed to oxidize it rather than reduce it!
Some thoughts:
Does your burner really need a wick? Would a properly sized orifice work just as well or better? In axehandle's thread I made reference to
industrial burners atomizing the molten sulfur, and that the S/O ratio is important for efficient burning.BeerChloride - 26-10-2006 at 18:19
not_important, where did you get your mp data? 100% sulfuric acid freezes at +10.4 C.
mericad, not_important is right about the outgassing. Your pan has to be a lot hotter than the temp you want the liquid to boil at. You have to have
"clean" materials. I like the idea of glass pieces for certain things when you want to be sure of purity and non-reactivity. Though contained air
works much better for boiling stones, glass does work (you may have to add 5 or more pieces). I made a simple retort from a glass rod and a broken
vacuum adapter, and found it fairly easy and painless to get to 87% from battery acid, but I noticed that there was a fairly heavy reflux going on in
the flask which did not make it through the retort. I would definitely hesitate to do an open flask boil-down.Fleaker - 26-10-2006 at 18:20
As far as I'm concerned, if it burns lean with more oxygen than necessary, it's of little significance since residual oxygen and any SO3 produced
aren't a bad thing. I'm just hoping that the V2O5 will get the job done. I want to avoid an orifice if at all possible--I'd much rather prefer a
sulfur "candle". Also, how big to make it? What's a feasible (and safe) amount of SO3 to produce? Questions like that still need to be thought
through. I'm looking at the wick part from the stand point of ease and reliability. Since I'm only keeping the sulfur about 10* above its melting
point, it might solidify in the orifice. With a wick, as long as the sulfur is a liquid, capillary action will be on my side.Rosco Bodine - 26-10-2006 at 20:31
I don't think a wick type of burner is going to produce enough SO2 output . Something I have considered
is using an old five gallon propane bottle as a combustion chamber . Cut a rectangular hatch door
in the side of it and hinge the cutout as a door piece
after pop riveting some added strips to its perimeter as a flange . Level out the floor inside the chamber with sand or vermiculite , and set in the
middle a ceramic radiant burner element from a gas space heater tilted at a fair incline ~45 degrees with its grid openings upwards , and the open
bottom plugged with about an inch thickness wad of kaowool . The kaowool could be soaked with xylene and ignited to preheat the element to a high
enough temperature so that molten sulfur dripped into the open
grid would inflame immediately and burn away as it flowed downward by gravity across the hot ceramic .
The door could be fitted with a mica or glass window
for observing the combustion and adjusting the sulfur drip until a smooth self sustaining burn rate is accomplished .......almost identically as is
the scheme
used for a waste oil burner .
The ceramic radiants can be gotten from old gas heaters or even ordered from places like eBay , like this one
Of course you will need a forced air supply and either a blower speed control and /or some sort of valve to regulate the air flow to what is optimum
for the drip rate of the sulfur .
The drip tube would have to be coaxially mounted inside
a much larger and heavier tube and well insulated right up to the tip from where it drips inside the combustion chamber to avoid excessive heating of
the molten sulfur by the combustion chamber heat , which would cause the molten sulfur to thicken and not to drip steadily . A proper sizing
for the molten sulfur feed line and the proper feed rate and temperature would have to be worked out during test runs
to find the optimum settings for everything . Once worked out , the burner should run okay unattended . The catalytic
reaction portion of the reaction could be also worked out so that the the reaction temperature is self-regulating at a particular feed rate from the
combustion chamber . A heat exchanger loop from the catalytic converter could be used
to help keep the sulfur molten in the reservoir with little or
no supplemental electric heating after the system is in operation , the heat energy from the burning and catalytic conversion being sufficient to
maintain the operation of the system . If feed air is being supplied from a compressor maintained supply tank , the air will be sufficiently
dehydrated already , as the water can be drained as
condensate from the bottom of the compressed air tank ,
and the regulated low pressure takeoff air from the high pressure supply tank will already be very dry , having lost its moisture on cooling in the
high pressure storage tank .
[Edited on 27-10-2006 by Rosco Bodine]evil_lurker - 26-10-2006 at 22:28
It would be expensive, but what about pumping compressed O2 directly into the sulfur burner?
Then having another oxygen inlet going into the reaction tube where pure O2 is pumped in preheated via a torch or whatever?Rosco Bodine - 26-10-2006 at 22:59
Air works just fine so why bother ?evil_lurker - 26-10-2006 at 23:13
I was thinking that
a) the sulfur would burn better and at a more easily controlled rate using pure O2... no need for a wick
b) since there is no moisture in the system, cheap iron pipe could be used with little corrosionnot_important - 27-10-2006 at 07:40
BeerChloride, you're right. I mistranslated 'pure' as meaning 100%, the book was using the 'pure' for the constant boiling not 100%. But then 100%
acid takes SO3 to get.
Burning in air actually gives more SO3, the small amount of NOx formed catalysis the conversion of SO2 + O2 to SO3, as in the chamber process. Burning
sulfur in pure O2 gives a fairly intense flame, try it on test tube scale sometime. Burns better, but controlled may be problematic
To avoid moisture, dry the air that feeds the burner. Use calcium chloride or silica gel.Nerro - 27-10-2006 at 07:56
Maybe a conversion of the lead chamber process would work, Mix your sulphur with nitrate and ignite by heating it for a while with a hot flame. (When
they're properly mixed you might even be able to with a few sparklers os something similar.) Lead the exaust gasses, (SO2 and NOx) through the pipe
with the catalyst or maybe even just a hot pipe, the NO2 will convert the SO2 to SO3 and the NOx should revert back to nitrogen (not sure though). The
SO3 can be led through H2SO4 to complete the synth.Magpie - 27-10-2006 at 08:34
When I was fresh out of college I worked at a pulp mill that used CaSO3 as a bleaching agent. The CaSO3 was made on site by first burning sulfur to
SO2. There was a view port to look at the burner. It appeared to be a series of jets that looked just like what you'd see in a furnace burning
natural gas. This makes me think that the sulfur was vaporized prior to burning. Now that seems like it would be controllable and trouble free, if
properly designed.Fleaker - 27-10-2006 at 12:49
Sounds like a reiteration of what I've said already. I understand the air needs to be dry, (well actually it doesn't matter if it's SS 316) and have
accounted for it. I guess the thing I'm most interested in is making the sulfur burner. I do not see why a large enough wick wouldn't produce enough
SO2. I guess what I need to do is saturate a piece of kaowool with liquid sulfur, wait til it cools, then mass to see how much sulfur loading there
is. I know that it's about a 2.5:1 factor for a liquid of 1.3 g/mL (experiments with other catalysts on this useful substrate): Per gram of kaowool,
two and one half times that of the liquid will be held.
Nerro's idea is interesting, but it might be hard to control. I'm afraid of over doing it and pushing way too much SO2 through the system and
overheating it.Nerro - 27-10-2006 at 13:06
It might be controllable by adding amounts of something inert like ground up silica to moderate the reaction. Experimentation with different ratio's
of silica might give you the correct amount to give you a steady but modest flow of exhaust gasses.
You might also spread the gasses through the sulfuric acid more effectively by placing an inert wool like material inside it and passing the bubbles
through that. It will break them up and disperse them while keeping their rate of ascent low.Eclectic - 27-10-2006 at 13:20
I may have mentioned this before:
How about a very small jet of O2 impinging on molten sulfur in an atmosphere of sulfur and SO2, followed by an afterburner? Has anyone tried it?Magpie - 27-10-2006 at 14:12
Fleaker I think I understand what you are saying with the wick. I'm not saying this, or any other method, might not work.
I'm just saying that upon reflection of what I think I saw many years ago the sulfur may well have been a vapor before it issued from the jets (which
was probably just a drilled pipe) and was burned.Rosco Bodine - 27-10-2006 at 16:32
I think the problem with any wick sort of burner is the
same as what would be the problem with just setting
an open pot of sulfur in the combustion chamber and letting it burn , and that is the temperature at which sulfur is fluid and free flowing will soon
be exceeded by the combustion chamber temperature , causing the
unburned sulfur to gell and thus making the replenishment of sulfur in the closed combustion chamber a problem while it is in operation . So any
design reliant upon molten sulfur in a fluid state as the
fuel feed for the burner is going to require some sort of reservoir which is controlled at a lower temperature
than the combustion chamber temperature in order for the molten sulfur to remain fluid enough and not become too viscous to feed the burner .
The molten sulfur is quite flammable so getting it to burn in the hot combustion chamber is no problem , and it
would likely burn very well simply dripped upon a small
mound of broken " lava rocks " like are used in gas cooking grills , if these were heaped in a clay saucer
like is used under clay flower pots as a drain pan .
Ever noticed how the melted fat burns on those lava rocks when it drips from a steak being grilled ? That
is the idea , and the burning of the fuel keeps the rocks
quite hot enough so that any new molten fuel dripping
upon them is also inflamed and the burning continues ,
with a continuous flame there on the ceramic material
acting as a burner and igniter for fresh material as it
arrives by drips . Increase the drip rate and you get a bigger fire , decrease the drip rate and the burner throttles back to a minimum sustainable
idle level .
So the combustion process is throttleable .
On a molten sulfur reservoir , the temperature will need to be thermostatically controlled , and the drain valve
and the exit tubing also will have to be wrapped with heating tape and thermostatically regulated so that the
sulfur remains at the most fluid temperature and can be flow regulated at a steady viscosity . And where that
feed line enters the much hotter space inside the combustion chamber it must be insulated against any undue heating which would thicken the sulfur and
prevent its flowing from the discharge . Indeed some
flow rate of the feed sulfur will be needed to keep this
last section of the discharge line free flowing and it
should be large enough bore so that it is self draining ,
perhaps even jacketed with oil or something which would
be temperature limiting for that final length inside the combustion chamber above the burner where heat
could complicate the flow of fuel to the burner . This
would be the logical location for the inlet air to the combustion chamber , coaxially flowing inlet air around the molten sulfur delivery tip would
keep it shielded
from the much hotter gasses of the combustion chamber .
Keeping the molten sulfur at the proper temperature
within the last few inches of the feed line is really the
most technically difficult hurdle of the entire scheme ,
but entirely manageable .
The scale of the burner and the rest of the apparatus
should be along the lines that could burn several kilograms of sulfur per day at a minimum , for any
practical amounts of product , otherwise what you
are doing is investing a lot of time and work and materials in a novelty which won't produce any worthwhile amounts in any reasonable time .
A simpler alternative if you want to manually feed sulfur
periodically to the burner is just to cast your molten sulfur
into short cylindrical ingots just slightly smaller than a metal tube which is mounted to deliver the chunks of solid sulfur to an open pan burner in
the combustion chamber . When the
burner gets low on fuel , you simply load some sulfur ingots
into the tube and push them through with a rod , where
they fall into the burner . It may not be high tech ....but it should work like a charm .
[Edited on 28-10-2006 by Rosco Bodine]Fleaker - 27-10-2006 at 16:57
Sulfur is cheap, 50lbs would be something like 10 dollars from the local farmer supply. I haven't even bothered to do the stoichiometry yet to see the
maximum (theoretical) SO3 output. I appreciate your advice Rosco, I think what I could do is set up some sort of thermostatically controlled sulfur
melter of perhaps 1 gallon in capacity and use a ball valve to control the flow rate. This would be in an elevated position and would flow down into
the reaction chamber. I like the concept of throttling it down or up, but I still recognize that additional dry air must be supplied to the combustion
chamber. I plan to also use a slightly faster 'jet' of air at the top of the apparatus (near the admittance point for the sulfur drip tube) that would
create a pressure differential in the bottom of the combustion vessel and thereby draw the sulfur dioxide into the reaction tube at a faster rate,
with oxygen at an excess.
Curious, but does anyone have information on catalyst service life (i.e how long before degradation occurs)?
ThanksRosco Bodine - 27-10-2006 at 18:17
I have been thinking about that open pot sort of burner
using a simple feed of solid ingots for replenishment from
a horizontal side tube , and there is a way to throttle
that sort of burner too by using a disc or a perforated
disc or dome to variably cover or expose the surface of the burning sulfur in the open ( or not so open ) pot .....
if you use the principle of a damper . This sort of scheme
could simplify the design of a prototype combustion chamber considerably and eliminate the neeed for any
thermostatically regulated molten sulfur feed systems .
You know how if you have a fire in the kitchen in a pan of hot oil , you simply put a lid on it to extinguish it ....
and if you had the lid center mounted on the end of a rod
so that you could raise or lower it gradually over the burning pot ....then you could throttle the flames across
a range of intensity . A cover plate could also be pivot mounted so that as it pivots it eclipses the open pot across the range of from fully open to
fully closed .
So there are a couple of ways an open pot burner could be throttled if you follow what I am describing .BeerChloride - 27-10-2006 at 19:01
I realize the going topic here is focused on the SO2 production, but here's some freezing data I compiled on H2SO4:
The mono- and tetra- hydrates can be identified in the graph. The dashed portion entails some estimation.
Now I know why my brown 94% didn't freeze. The impurities probably lower it even further (guess - 45 ?). Someone asked where to find dry ice. It's
available at the grocery store here (surprising, I know), but it should be able to be found at food/catering supply establishments. I'm getting rather
curious to try freezing.
Dry ice is -78 C, so it should work. What should I do? Acetone and dry ice? Does acetone simply provide heat transfer, or are there other reasons for
this mixture?mericad193724 - 28-10-2006 at 06:20
BeerChloride, Maybe you should try to dilute you brown H2SO$ a little bit so it is in the 85% range, according to your graph it would be much easier
to freeze out, maybe even with a standard freezer!
Doesnt look like my "battery acid boil down" is going to materialize today...its raining and very windy so I would have to do it in the closed
garage...BAD IDEA!
Mericadnot_important - 28-10-2006 at 07:16
Quote:
Originally posted by mericad193724
BeerChloride, Maybe you should try to dilute you brown H2SO$ a little bit so it is in the 85% range, according to your graph it would be much easier
to freeze out, maybe even with a standard freezer!
Mericad
That would be the mono-hydrate freezing out, You'll not get any more concentrate acid than that by freezing once you take to the 85% or lower range.BeerChloride - 28-10-2006 at 07:50
Yeah, it crossed my mind to dilute it, but that extra concentration above 85% is precious - where the real dehydrating power is. But, it could still
be a way to purify it in order to have some not-so-concentrated stuff. Incidentally, the 94% might be close to eutectic, I don't know, but I don't
expect the freezing to concentrate anything. I'm just aiming to purify and keep the 94% concentration.
I meant to get some dry ice earlier and sublimed some iodine instead...mericad193724 - 28-10-2006 at 17:21
This maybe a really stupid comment but....
Can't you just do electrolysis on a sulfuric acid solution to drive off the water as H2 and O2. This can just be led off into NaOH or outside. This
setup can be run over night and maybe produce 90%+ from 30% acid? Eventually the conc. acid would just act as a short circuit so you should include a
fuse to blow and then you got conc. acid right?
Will this work, I will try if it is a yes...This seems safer than boiling
MericadThe_Davster - 28-10-2006 at 17:38
You will form sulfurous acid, oxygen(with small % ozone, this is actually a prep for dilute ozone in oxygen gas mixtures), hydrogen(most likely, but
the redox potentials don't completly indicate this), and water.
[Edited on 29-10-2006 by The_Davster]BeerChloride - 28-10-2006 at 17:47
Good try, but won't work. The reduction potential is higher for the H+ to be turned into H2, rather than from water. Thus, the acid will be destroyed.
Many electrolytic processes drive the pH up at the cathode, and can be a way to make hydroxides. I saw a thread somewhere in here, though, that may
have mentioned making H2SO4 by electrolysing magnesium sulfate or something.
Just another thought: another possible variation might be to concentrate batt. acid to about 87%, which should freeze out monohydrate around 0 deg.,
the remaining liquid will be more concentrated.unionised - 29-10-2006 at 02:07
"Good try, but won't work. The reduction potential is higher for the H+ to be turned into H2, rather than from water. Thus, the acid will be
destroyed."
What will the acid be converted to?
Turning H+ into H2 (and OH- into O2) is exactly what we want to do here.
As far as I can see this method will work to produce relatively conc acid from dilute stuff (albeit slowly and expensively).
Unless the nascent hydrogen reduces the HSO4- to SO2 I can't see a problem and, since car batteries outgas H2 if they are overcharged, I don't think
that reduction will happen (at least not in solutions as concentrated as battery acid).
What happens in really conc H2SO4 might be different, but I think this should work.
For what it's worth, the same idea (electrolysis) is used to dehydrate HF because most drying agents don't work.BeerChloride - 29-10-2006 at 05:11
Well, unionised, I just don't know. What the_Davester said sounds plausible. But you raise a good point about car batteries, and I read that they give
off hydrogen AND oxygen when overcharged due to electrolysis of water. To me it makes sense, though, that the cathode will put electrons into the H+
(properly H30+) ions which will then combine into H2 leaving water. Oh, wait, the concentration dependence! A steady-state equilibrium. For dilute
acid it might work, but at a certain point of H+ concentration, the H2 production balances. That is why the overcharged battery bubbles, and the acid
cannot be concentrated any further. I think the H2 comes from the acid.Nerro - 29-10-2006 at 08:20
I remember vaguely that I once read about producing H2O2 by electrolysis of sulfuric acid. Perhaps that might also be formed.The_Davster - 29-10-2006 at 10:26
With HF it works because look at the potentials:
F2 +2e- <-->2F- +2.87V
O2 + 4H+ +4e- <--> 2H2O +1.23V *
2H+ +2e- <--> H2 0 *
2H2O +2e- <-->H2 +2OH- -0.83
So When electrolysing HF, you get a net of:
4H+ + 2H2O --->H2 + O2 +4H+, so water is removed as you said.
However with sulfuric...
O2 + 4H+ +4e- <--> 2H2O +1.23V *
SO42- +4H+ +2e- <-->H2SO3 +H2O +0.137 *
2H+ +2e- <--> H2 0
SO42- + 4H2O +6e- <---> S(s) +8OH- -0.751
SO42- + H2O +2e- <---> SO32- + 2OH- -0.936
2H2O + 2e- <--> H2 +2OH- -0.83
So when electrolysing sulfuric:
2SO42- +8H+ +2H2O --> 2H2SO3 +2H2O +4H+ O2
Will aproximatly happen, because the acidic sulfate is a better oxidizing agent than H+, then what happens with HF will not happen here. But, based
on the number of preps for preparing everything from ozone to oxone from sulfuric electrolysis, a lot of other products may form depending on the
conditions, but one thing is certain, that the water will not just be electrolysed out.unionised - 29-10-2006 at 13:21
Last time I checked the electrolysis of dilute sulphuric acid to give H2 and O2 was a standard high school demonstration (as well as a common
observation with overcharged batteries).
If you tell me that it doesn't work, because the ionisation potentials say so, then I say you have got something wrong.
The minor technicality that you are quoting standard oxidation potentials whereas the real reaction we are talking about is miles from the standard
conditions is one point that may explain this.
More to the point, if the H+ is removed from the acid or the water it doesn't matter- the mixture will re equilibrate. If the system loses H2 and O2
then the overall effect is loss of water.
You also seem to have come up with a cell reaction that doesn't make sense- peroxide oxidises sulphite rapidly so they can't both be the products of a
real reaction.
Since, as has been pointed out, ozone and H2O2 are minor products the overall mixture will be oxidising- S(IV), if formed, has a fair chance of being
reoxidised to S(VI).
Peroxydisulphate is another reasonable product, but it's not much of a problem it breaks down to H2SO4 and "O"The_Davster - 29-10-2006 at 15:56
In the standard electrolysis demonstration, to be honest, I do not know why the sulfuric does not get reduced first, and then proceed to just
electrolyse the water. I had always been told the sulfuric is just there to increase the conductivity of the water. But this is at condidtions where
the sulfuric concentration is much below the standard 0.1M
Ah, I realize the discrepancy, the sulfate being reduced to sulfurous, came from a not-so-reputable book, a high school level text, wheras my
analytical text has no mention of this reaction. It was likely as you mentioned an oversimplification of a nacent hydrogen reduction. The 2H+ +2e-
<--> H2 reaction is next in line, which does support that you could drive off water by electrolysis, but at 0.1M. If I were so inclined, I
could dig up my old notes on how to convert potentials at various concentrations....but...I am not in the mood for all that mathemagic now.
But all sorts of other, less predictable side reactions will still occur, so I say that it is worth a shot to see what will happen, the elimination of
water may or may not be the primary reaction, but after you try it, be sure to do some analysis on it, and the gasses formed.mericad193724 - 29-10-2006 at 18:02
I think I could probably try this out. I got a battery charger that can do 12V or 6V at 10 amps or 2 amps. The liquid will probably be very conductive
and get hot upon electrolysis. I think carbon rods would dissintergrate very quickly in these conditions.
What type of electrodes should I use for this test? I will start with a small scale electrolysis around 50ml 30% acid.
Mericad12AX7 - 29-10-2006 at 18:04
I've electrolysed sodium sulfate with lead anode, don't know how graphite would fare. The lead oxidizes to a thick (strained, somewhat flaking) layer
of PbO2 so a heavy bar of lead would be prudent. Don't know about acid.
TimRosco Bodine - 29-10-2006 at 22:50
Electrolysis has come up so often lately that I am
building a transformer based 40 ampere *continuous duty* variable 0-15v. DC supply from scrounged parts .
So far I have the variac , and the fixed step down transformer , the bridge rectifiers and heatsinks ,
a 4.5 inch 50 millivolt scale precision meter , which
will be across a .001 ohm solid 6 gauge 29.25 inch
copper shunt for reading amperes .
The plan is to use round jacketed three conductor 12 gauge extension cord for the added flexibility for the
output cables , and connect the three conductors
" three in hand " to give a shared conductor of
over 8 gauge and slightly smaller than 7 gauge size ,
to provide cool running and low loss output cables .
The terminal anode clamp contact is going to be silver , and heavy duty even if I have to fabricate it from bullion .12AX7 - 30-10-2006 at 08:14
Just pick up 10-20 feet of stranded 8AWG at the hardware store... and for the shunt, use a few inches of bare #10 or #12, a miliohm is only
dissipating 1.6W at full rated current...
TimRosco Bodine - 30-10-2006 at 10:11
Looked at the 8 gauge stuff but it is too stiff , meant for
permanent wiring and not for flexible hookup . I want something soft and flexible like welders cable . The specialty stuff is special order only
and it is a lot more expensive than than just getting a heavy duty extension cord and doing what I described going three in hand
on the conductors .
And as for the shunt , the size is common panel ground
~3/16" bare copper and has heavy machined copper T-connectors that slide for positioning like a split set collar to provide easy adjustment for
calibration and solid connection . I figured on just sleeving the shunt with
tygon tubing or split loom sleeve insulation and bending it into a wide U shape fastened to the base of the power supply . It was simply chosen
because it was the smallest size where all the hardware for needed connections was commonly available off the shelf , even though it is heavier than
necessary I know . There won't be any thermal drift on 1.6 watts due to conductor heating across 29.25 inches of 6 gauge copper .
The two transformers alone weight about 35 pounds ,
and everything is passive air cooled ...no fans .
I am going to add a polarity reversing switch for electrode preparation , voltmeter , self-resetting overcurrent breakers ( at least ) and possibly a
thermal breaker and fuses for backup protection ....since this thing will be run unattended for perhaps days at a time .
I have a 20 gallon aquarium which I keep thinking
looks a lot like a perchlorate cell to me
I have a couple of 12 liter pyrex jars with 100mm threaded ring and disc cover assemblies that could
be put to use too .
You know a cell running at low voltage like 3 or 4 volts
and high current , is really a scenario where a center
tapped transformer and a dual Schottky Diode bridge
is much more efficient , in comparison to a single output transformer and full wave bridge rectifiers having four
standard diodes . I could never seem to scrounge a
good center tapped transformer having a high current secondary at a cheap price .....so I have the cheaper and less efficient solution .
But I did save a link for some high current Schottky devices for cheap , in case anyone else might be interested in such a component for improving
rectification
efficiency greatly on low voltage high current DC power supply project .
I would use a couple (three) schottky diodes pulled from computer supplies. In the 300W range, these are typically 40A, 30V dual (common cathode) in
a TO-3P or TO-247 package.
TimRosco Bodine - 30-10-2006 at 11:07
Yeah I really wanted to go with Schottkys , but I didn't want to buy extra heatsinks and micas , and having
no center tapped transformer meant I would need
four of the damn things ..... more than twice as efficient
as standard bridges and heatsinks I already had on hand .....so I stayed with what I got .
The four 35A monolithic full wave bridge rectifiers are
mounted in isolated bare metal square cases , which
I am mounting back to back in paralleled pairs in the
web of two separate 50W H-finned heatsinks , wiring
all four in parrallel to handle the heat dissipation and
avoiding having to use the expensive massive or fan cooled heatsinks ....by spreading the load comfortably
enough across what would be a 140A bridge array if it
was more massively heatsinked . Cost about half as much to afro engineer it the way I have got it
Heatsinks are damn expensive aren't they if you aren't
buying them by the ton .BeerChloride - 31-10-2006 at 06:37
Interesting plan, Rosco. Yes, I often try to avoid all but the simplest heat sinks. My largest PS is a homebrew 12 v, ~10 A dual transformer,
pseudo-regulated with 2N3055's. I need to make a variable one for at least that kind of current.
I wanted to post about dry ice. Finally got some - more dry ice than I've ever seen. Now don't get me wrong, I used to play with liquid nitrogen all
the time, so CO2 pales in comparison, but it's neat too. Big 10 lb block. I used a wood chisel and a hammer to break it up. BTW, acetone with dry ice
is great - poor man's liquid nitrogen! (actually LN2 is just as cheap) It fizzles away the dry ice at first, then becomes totally stable with no
"clouds" like you always have with LN2, and almost no odor. After a while I decided to touch the cold acetone - it just looked so much like icewater!
My finger made a sizzling sound when I put the tip of it in - I had my finger out already before those auditory impulses even reached my brain. I DO
NOT recommend doing this! I am a trained mad scientist. Well, actually I am a trained scientist. So I shouldn't be doing such things, either.
I chilled several ml of the brown H2SO4. It became extremely viscous to the point of barely flowing, but didn't freeze after several hours. The 94%
seems to behave similar to sugar perhaps. Sulfuric acid rock candy???unionised - 31-10-2006 at 10:02
Err, is any of this chemistry?not_important - 31-10-2006 at 10:15
It relates to making strong sulfuric acid, so I think it's still on topic.
Low temperature freezing seems to readily give glasses or gels instead of crystals. The liquids get viscous enough that crystallisation is slowed
down, very slow cooling and extended times are needed for proper crystal growth. It might prove difficult to crystallise sulfuric acid of this
strength.
I use fairly dry ethanol or isopropanol instead of acetone with dry ice. A little less flammible, and less agressive at dissolving organics so I can
use plastic containers and tongs. However I believe that acetone gives you a bit lower temperature, I don't know why that would be.
The bath liquid serves several purposes. One is indead a heat transfer meadium, to get good contact. Another is temperature control, with liquids with
freezing points above the temperature of dry ice you can make a slush bath with a temperature near the freezing point of the liquid. There seems to
be another effect, as a number of books list different temperatures from dry ice in differing liquids that don't freeze at -70 C. Dissolution cooling,
perhaps?mericad193724 - 2-11-2006 at 13:00
It turns out electrolysis of dilute H2SO4 will not concentrate the acid, it will form ozone as Davster said....
this is according to wikipedia:
Laboratory production
In the laboratory ozone can be produced by electrolysis using a 9 volt battery, a pencil graphite rod cathode, a platinum wire anode and a 3M sulfuric
acid electrolyte. The half cell reactions taking place are:
So that in the net reaction three equivalents of water are converted into one equivalent of ozone and three equivalents of hydrogen. Oxygen formation
is a competing reaction.
This is only at 3M H2SO4.
Mericad12AX7 - 2-11-2006 at 14:53
S'still removing oxygen and hydrogen...why would you say it wouldn't concentrate it?
TimRosco Bodine - 2-11-2006 at 17:52
Could it possibly be forming peroxydisulfuric acid ?12AX7 - 2-11-2006 at 18:11
Done hot it would tend to decompose, no?
How about distilling it when you're done?
Oooh, gaseous H2S2O8...toasty
TimRosco Bodine - 4-11-2006 at 08:13
Know it is off topic , but regarding the linear power supply for an electrolytic cell or plating tank , I am
looking at doing some sort of basic voltage regulation
which is not terribly lossy nor requiring huge heatsinking
for any regulator dissipation , and I am thinking that
simply using a sizable capacitor filter may be adequate
for the task . Or if it doesn't do the whole job , it will
take a lot of the ripple out and ease the job for any regulator stage downstream . On the 40A transformer
output , I am considering using four 35,000 uF 25V caps paralleled across the output of the bridge for the main filter .
I haven't done any calculations on this filter capacitor value and am open to suggestions .12AX7 - 4-11-2006 at 08:57
2000uF per amp is a rule of thumb. Best around 12V though. The rule of thumb should really say 24m F*V/A (that's m = mili), since ripple voltage is
independent of supply voltage but %ripple depends on supply voltage. This would suggest as much as 0.2F for your supply, which should be made of a
number of paralleled capacitors as you will draw a pretty good ripple current.
Instead of capacitance, you may consider inductance instead. Some 6-8AWG around a couple MOT cores, suitably gapped, should provide enough inductance
to filter your load with little or no capacitance (none needed on the upper slope of the cell's exponential curve, but low voltage or current loads
will need capacitance).
TimRosco Bodine - 4-11-2006 at 10:43
I am using a variac to vary the AC on the primary of the fixed transformer , so that I can drop the secondary
voltage without dissipative losses any more than
necessary across any regulator . It would be great
if I could get the ripple as low as possible with low loss
filtering , and not have any active regulation stage at
all , essentially flattening the 120Hz pulsed DC to
as close as possible to its steady state rms value ,
by a brute force filtering approach .12AX7 - 4-11-2006 at 12:19
Well, with a 10H choke of 1mohm resistance followed by about 100F filtering will certainly get a wonderfully smooth DC output, if you don't mind the
energy storage. Indeed, the ripple attenuation approaches infinity as L and C approach infinity. That much is obvious, and it follows that ideal
attenuation cannot be had in a finite universe.
Thus, the question is not "can I get the ripple as low as possible?", but rather it must be "how low can I get ripple, economically?".
Interestingly, you can also use phase control, instead of expensive variacs, to vary the output voltage when choke-input filtering is used.
Electrical control can then be added, allowing for electronically regulated response.
Timunionised - 5-11-2006 at 05:51
To get slightly closer to the topic again; how much does the ripple matter to electrolysis?Rosco Bodine - 5-11-2006 at 09:16
Suppose you have an electrolytic reaction which
may produce any of several different products depending
upon the applied voltage to the electrodes , and there is
only a couple of tenths of a volt variation which will cause
different materials other than the one desired to be the product . The swinging value of the voltage from ripple
could cause all the possible products to be produced in
sequence at the frequency of the ripple voltage , so you end up with a mixture of products instead of the one product for which a steady state and
specific voltage is required .
For many electrolytic reactions , an unregulated DC may
not cause any problem other than perhaps increasing
evolution of gas from the electrodes at a lower average
current , thus decreasing the efficiency of the cell .
But for reactions which are voltage sensitive with regards to the products possible .....the effect of a varying voltage could be disastrous to yield
by producing
unwanted byproducts at the same time as the one
product desired . This not only wastes precursor material
but may then complicate isolation . And if the different
products are reactive with each other , it could render
the hoped for reaction impossible all together using an
unregulated supply .
From what I am seeing too in the transformer manufacturers design recommendations , most are
saying to derate the AC current specified for a
transformer to which a full wave bridge rectifier and large capacitor filter is added by a factor of 1.8 .........which
really stinks because what that means is that is the
rectified and filtered DC output current available for regulation before it is even regulated , is only 55.5%
of the AC current rating of the transformer and that is
a huge bite , when a 100 Amp AC secondary is basically
only good for about half that current after the rectifier , filter , and regulator are added . And it is a fair bet
that the " lost efficiency " is dumped as heat which
adds another problem of how to dissipate that wasted
energy .
Switching supplies do better , but still have quite a bit of loss in their circuitry , the main advantage seeming to be
reducing component size and weight for working at higher operating frequencies .
From what I have seen looking at low voltage high current supplies in terms of efficiency and bulk , for
any sizeable electrolytic cell like a commercial reactor ,
on site generation using specially wound DC generators
and brush commutated DC would absolutely rule in
terms of economy of operation . These would be
driven by electric motors , or diesel or steam or hydro .
From the looks of it , when more than a couple of hundred amperes of DC requirement are presented ,
the engine driven DC generator will rule as a source
of power . And next to that , multielement polyphase AC alternators driving Schottky rectifiers , having their elements outputs incrementally phased
, could provide
a synthetic DC having a high enough frequency and
low enough ripple voltage that filtering alone would
suffice .
If a serious high current and * high efficiency * DC source is required , and what I understand at this point is
correct , it really needs to be done from the actual power generation stage up , and be addressed as a power generation project , not as a power
supply project which is simply plugged into the existing AC commercial grid .
Anything in the way of a bench size linear supply with filter and regulator which can be easily enough built from off the shelf components is probably
going to reside in the range of
10 to 50A output , and weigh about a kilo per Ampere for
the size of the cores , case and other components .
[Edited on 5-11-2006 by Rosco Bodine]unionised - 5-11-2006 at 12:08
So, unless you set up a potentiostat which monitors the working electrode's potential with respect to a reference electrode (Ag/AgCl is popular) it
won't work properly.
Of course, all those reduction potentials are concentration dependent so, not only do they change as the reaction goes on, but the depletion of the
reactants (and the buildup of products) in the vicinity of the electrode also messes things up. At least you need a good stirrer.
Fortunately, for the reaction we are looking at 2 H2O--> 2 H2 and O2, this doesn't matter much. If you ramp up the current density far enough then
you can get ozone (though some of the patent data indicate it's bloody hard to get better than 50% yield of ozone).
When the voltage drops then the current falls dramatically- you get a lousy waveform but I don't think it's worth producing a regulated smoothed
supply for stripping water from dilute sulphuric acid.Rosco Bodine - 5-11-2006 at 12:37
Long before you get to the point of having concentrated
H2SO4 from water loss by electrolysis , what you will be doing is making persulfuric acid .unionised - 5-11-2006 at 12:57
That's OK by me. I get rid of hydrogen by electrolysis; some of the excess oxygen gets incorporated into persulphuric acid and some leaves as
ozone/O2.
I add a little Pt as a catalyst to decompose the persulphuric acid and I've got conc H2SO4.
I think the really big problem here is the number of amp hours it takes to get rid of a litre of water.
A litre is about 56 Moles of H2O so I need to convert about 112 Moles of H+. The Faraday constant (IIRC) is about 100000 Coulombs per mole so I need
to get 11200000 Amp seconds of charge through the stuff. Something like 30 amps continuously for 4 days. (could someone check that please).
I'd be impressed by the electrodes that stood that.
Suddenly distilling out the water looks like a really good idea.Rosco Bodine - 5-11-2006 at 13:06
Does the process go all the way to oleum ?
And what sort of voltage increase is experienced due to
the drastic increase of resistance as all water disappears
from the H2SO4 ? Isn't it something of a contest to see
which will happen first , electrolysis or actual boiling of the acid ?
[Edited on 5-11-2006 by Rosco Bodine]12AX7 - 5-11-2006 at 13:16
Doesn't persulfuric acid decompose on heating and form much better at low temperatures? How about some Mn? (Even better: use an MnO2 anode!)
The reason switching power supplies are so pervasive today is 1. they are much lighter and cheaper (materials and labor) than iron and copper power
transformers, and 2. they are far more efficient at producing a regulated voltage than a corresponding linear regulator.
Most SMPSs use a cap-input filter, giving them bad power factor. Some use PFC (Power Factor Correction) chips to provide a pre-regulated high voltage
supply without all the nastiness.
Power factor is NOT loss. Indeed, that it is imaginary "power" is exactly the reason it is a concern! The reason is, capacitors are only charged
during the voltage peaks. This causes huge spikes in current, leading to I^2R loss in the wires (from the rectifier and filter, through the
transformer, all the way back to the generator station). By integrating, it can easily be seen that this piecewise current draw dissipates more power
in the wiring than the same average current would if drawn by a pure resistive load. A transformer has limited heat dissipation capacity for its
wiring, so a limited amount of RMS current can be drawn from one. Since RMS current is higher (per DC current output compared to a resistive load),
the current output must be derated.
A choke input filter, as suggested, has a constant-current characteristic (when current draw is greater than the critical value). This will much more
fully utilize the available current output of the transformer (and associated wiring), allowing nearly the full rated output capacity.
And BTW, generators have slip-ring losses, wiring and other BS. Don't be a fool. The industrial answer is always to connect many cells in series
anyway.
Tim
[Edited on 11-5-2006 by 12AX7]Rosco Bodine - 5-11-2006 at 13:36
Switching supplies are well done little pieces of engineering ,
but they are highly specialized too . You don't just grab the parts off the shelf and toss one together , at least not as easily as a line frequency
linear supply . The transformers
are ribbon wound ferrite core for example , and it just gets worse from there . So if you are manufacturing ten thousand units ....then build a
switching supply , most definitely . But if you are custom building one unit for personal use ....build a linear .
An yeah , a plurality of cells in series combined with polyphase rectification would be the industrial solution , along with building the
electrolysis plant on the river right next door to the power plant .
And brush losses are virtually nothing compared with
semiconductor losses , even Schottky rectifiers are
way more wasteful than a good old graphite brush on a DC generator ......
Precisely why they use brush commutation on locomotives
and other high current switching , even in the age of semiconductors ......some switching is still better done using
brushes . Vacuum tubes still rule in some areas too .
[Edited on 5-11-2006 by Rosco Bodine]12AX7 - 5-11-2006 at 15:47
Odd, I have the parts literally on my shelf. Including removed ferrite transformers, and various ones that I've since rewound. If I have suitable
ferrite, copper and MOSFETs, I would rather build an SMPS than a linear supply, because I don't have any transformers of that size around and damned
if I'm going to cough up a couple hundred bucks for one!
TimRosco Bodine - 5-11-2006 at 17:07
You must have an interesting junk box .....but
how do you even know what is the rating of the
ribbon wound ferrite cored transformers that you
cannibalize from switchers that are proprietary
parts ? They don't exactly provide the details
on all those " no user servicable parts inside "
that we all love to reverse engineer and try
to guess about . Is it hook it up and try it ,
do the old burn / no burn test ?
Hey I found a little line filter choke which caught my eye .
Light as a feather no doubt .
About a dozen of these in parallel out to give decent filtering on a 20 Amp linear supply , of course a forklift
will be required for moving the power supply , unless
it was rail mounted , you know the rails like a train
travels along May as well simply use a box car for
the cabinet .12AX7 - 5-11-2006 at 18:36
Doesn't look much different from about two MOT's worth, where MOTs are approximately free, as opposed to upwards of $50. That is a nice inductor,
though!
You'd be suprised how many schematics there are for commercial power supplies. But that doesn't bother me as the specs for a power supply such as
this are proprietary anyway. Is it really that much trouble to use some copper wire or strap and masking tape to make your own four-turn transformer?
TimRosco Bodine - 5-11-2006 at 19:42
Basically what you have with a switching power supply
is a VLF transmitter from which you have disconnected
the long wire antenna , and rectified and filtered the output .
In fact if you were to be creative , you could probably attract a whole lot of government attention with a bit of amplification
and off label use , of an ordinary switching power supply ,
because it would play hell with the VLF band that is monitored to the depths of the deep blue sea for submarine
communications . A sweep frequency oscillator in that band
would be like an invitation for black helicopters .
[Edited on 6-11-2006 by Rosco Bodine]12AX7 - 5-11-2006 at 21:49
That was...rather non-sequitur, unless you were trying to get at something, RFI perhaps? If your concern is RFI, please just say so.
If you're making an analogy to transmitters, please don't; the squarewave output of any switching supply would be illegal in any country, transmitted
at any noticable power. Harmonics must be a whole lot better than -3dB below the fundamental! Conversely, radiotransmitters (except possibly AM
transmitters, which aren't as efficient as a straight class D amplifiers) are not made to supply the kind of load (transformer and rectifier) demanded
of in switching supplies.
Also, the wavelength is such that very little LF or VLF would be transmitted. Much more likely is switching transients (from rise and fall times
under a microsecond) radiating into the tens of MHz region, which again can be attenuated with proper shielding and filtering practice.
Lastly, it takes a badly designed SMPS to produce a frequency sweep. They are PWM, not FM.
Tim
chamber!!
SAM4CH - 27-1-2007 at 06:11
What about the best material for combustion chamber? I tried to use stainless steel 316 but it was not very good!! I faced expansion in chamber and
there are some very very small pores in it!!ac- - 10-6-2007 at 08:00
Hope this isnt posted, but just saw this on wikipedia, "The aqueous solution on boiling loses some ammonia and forms an acid sulfate." http://en.wikipedia.org/wiki/Ammonium_sulfate
Wouldnt this be a good way for sulfuric acid?Aqua_Fortis_100% - 11-6-2007 at 07:21
Quote:
from the above patent:
(...)By this method it is achieved to decompose ammonium sulphate to a fully extent into ammonia gas and sulphuric acid liquid, where the ammonia gas
during processing is evaporated and separated out from a restsolution consisting of sulphuric acid.
This is interesting..i think which this had been discussed before on this board(and the pre conclusions were that the ammonium sulphate is decomposed
(if heated alone, without H2SO4 covering it) into NH3 and H2SO4 which recombine in the air making a large white and irritating smoke (almost as same
which happen to heated ammonium chloride).
But this seems to be strange: this this really a feasible method to make (or recovery) sulphuric acid?
seems to me that heating ammonium sulphate, in anyway, lead this to decompose (chiefly) into NH3 ,and H2SO4 (and further H2O and
SO<sub>x</sub> ).. but heating ammonium sulphate into conc. H2SO4, i'm considering which can be possible..but , is the resulting sulfuric
acid a pure substance ? i assume which at least some of this NH3 (re)react which the sulphuric acid to form again the ammonium sulphate... but in
these conditions , i'm not really sure..
have anyone tried this?
thanks
edit: thanks ,ac- , by provide a such patent...some months ago i iniciate to searching on sulfuric acid subject, and certainly i will thrown this
also to my "drawing search board" .
[Edited on 11-6-2007 by Aqua_Fortis_100%]not_important - 11-6-2007 at 08:38
The H2SO4 tends to oxidise the NH3, plus there is some decomposition of H2SO4 to H2O and SO3. The mix of SO2, SO3, H2O, and NH3 does make 'smoke',
what the gas+particulate phase composition is depends on heating and pumping rates. If you are pumping the NH3 off it does reduce the amount of
decomposition, but the yields still didn't seem to be that great.kelaniz - 30-7-2007 at 16:00
Interesting thread. At the risk of my first post here being stupid, I have a question.
How quickly does a solution of H2SO3 oxidize to H2SO4 (if at all)?.
I've seen this recommended a few times as a viable and (presumably) safer method of producing H2SO4, i.e. bubbling SO2 into water instead of SO3. Yet,
nobody seems to do it, so I'm wondering what the catch is.
-Kel
[Edited on 30-7-07 by kelaniz]12AX7 - 30-7-2007 at 17:04
Oxidizing it isn't too great I guess. Certainly works in the environment (SO2 from burning things and volcanos is the primary source of sulfate
aerosol), but that's a far cry from the conditions and the patience a mad scientist might be up for.
Timkelaniz - 30-7-2007 at 21:50
That makes sense. After all, acid rain doesn't decompose a car overnight, so it can't be that efficient a process.
Thanks for the reply! Any info from someone named after my favorite combo of 6AV6 triodes gets automatic credibility with me.
-Kel12AX7 - 31-7-2007 at 13:35
You're welcome.
Don't take acid rain lightly though -- you might study Sudbury, Ontario before the 1970s.
Timakbarnejad - 12-11-2010 at 09:13
Manufacturing of sodium sulfate from “salt cake”
The Mannheim process is a well known process for manufacturing of sodium sulfate from sulfuric acid and salt as:
NaCl + H2SO4 NaHSO4 + HCl
NaHSO4+ NaCl Na2SO4+HCl
As the second reaction is done in very elevated temperature, in some factories that HCl is the main product, they do not raise the temperate as needed
to complete the second reaction because of high corrosion problems. The solid from these furnaces is about 50% Na2SO4 and 50% NaHSO4 known as salt
cake.
I tried to put this salt cake in direct fire to convert the NaHSO4 to Na2SO4 as follow:
2 NaHSO4 Na2S2O7 +H20
Na2S2O7 Na2SO4+SO3
The problem is that the SO3 does not evolve from the salt cake unless severe mixing which is not practical in direct fire system and the second
problem is the absorption of hot SO3 mixed with hot fuel gas. The gas is very pollutant and absorption or distillation is very hard because the high
temperature.
I tried heating salt cake for 5 hours in 700 C but the sodium sulfate content has risen just from 50% to 87%.
Can anyone help me with this problem? Is there any other way to recover sodium sulfate from salt cake?
Thank you
Interesting thread. At the risk of my first post here being stupid, I have a question.
How quickly does a solution of H2SO3 oxidize to H2SO4 (if at all)?.
I've seen this recommended a few times as a viable and (presumably) safer method of producing H2SO4, i.e. bubbling SO2 into water instead of SO3. Yet,
nobody seems to do it, so I'm wondering what the catch is.
Sulfur dioxide is oxidized to dilute sulfuric acid in aqueous solution using air as oxidant and iron or manganese sulfate (better yet, iron and
manganese together) as catalyst.
The bad news is that none of the tests went higher than about 15% sulfuric acid concentration, though it should be possible to go a bit higher, and
the resulting acid is of course mixed with metal sulfates which will need to be plated out or otherwise removed before the acid is used for most
purposes*.
The good news is that the process uses very inexpensive and very widely available materials. It operates at low temperature and atmospheric pressure.
Although the Bureau's laboratory and pilot plant studies used special equipment to optimize conversion efficiency and rate, it is easy to imagine
performing essentially the same chemistry with dirt-cheap equipment, and on a fairly large scale.
*But not for the purposes the Bureau of Mines considered, where metals were already ubiquitous.hissingnoise - 4-6-2011 at 05:30
Quote:
The bad news is that none of the tests went higher than about 15% sulfuric acid concentration, though it should be possible to go a bit higher, and
the resulting acid is of course mixed with metal sulfates which will need to be plated out or otherwise removed before the acid is used for most
purposes*.
A hard look at all the methods described brings one to that inescapable conclusion --- there's always a catch!
All that's required, however, is absolute desperation and total commitment . . .
The bad news is that none of the tests went higher than about 15% sulfuric acid concentration, though it should be possible to go a bit higher, and
the resulting acid is of course mixed with metal sulfates which will need to be plated out or otherwise removed before the acid is used for most
purposes.
Since you mentioned using dirt-cheap equipment, this seems like an interesting process to develop
a solar still for, to reduce monetary inputs even further. I'm convinced that one of the good uses for solar power is for direct process heat for
industrial processes, and this looks like an interesting starting point.AJKOER - 12-6-2011 at 13:10
One way (which I have yet to perform, but its on my list) is to add sulfur (buy flowers of sulfur in the garden section and wash with distilled water)
to freshly prepared HClO (add vinegar to bleach and distill half off of the solution, which is 5/6 of the total available HClO vapor and the gaseous
anhydride of HClO, namely Cl2O. Repeat to further concentrate if needed).
Hypochlorous acid (HClO), per Watt's Dictionary of Chemistry, not only will oxidize (even dilute HClO) sulfur to H2SO4, but P to H3PO4, I to HIO3,
etc.
By adding H2O2 to HClO, you get HCl and O2. Heating HClO to around 70 C (or, let standing in sunlight) makes HCl and HClO3 via a disproportionation
reaction (actually, a two stage reaction that forms HClO2 first). Chloric acid, HClO3, is of course, the parent of all chlorate salts.
By the way, in most H2SO4 reactions, you can replace H2SO4 with NaHSO4 and run a safer reaction! So if you have NaHSO4, I will recommend you using it
before handling, boiling or exposing your lungs to SO3 fumes from any H2SO4!aliced25 - 12-6-2011 at 13:42
Interesting thread. At the risk of my first post here being stupid, I have a question.
How quickly does a solution of H2SO3 oxidize to H2SO4 (if at all)?.
I've seen this recommended a few times as a viable and (presumably) safer method of producing H2SO4, i.e. bubbling SO2 into water instead of SO3. Yet,
nobody seems to do it, so I'm wondering what the catch is.
-Kel
[Edited on 30-7-07 by kelaniz]
Extremely well if you add a halide (such as chlorine/bromine/iodine). The addition of SO2 to water is endothermic and the amount of SO2 that will
dissolve will surprise the piss out of you. Also, if you freeze, thaw, freeze, thaw, freeze & thaw the SO2/H2O solution and check it with litmus
each time, you'll note that the solution increases in acidity each time (the idea comes from a Japanese journal article [very little engrish] which
describes the reaction with dissolved oxygen) as the H2SO3 is oxidised to H2SO4.
Personally I prefer the halides, it is the only way I know of to make pure HCl/HBr/HI (not that I make HI that is illegal here) with pure sulphuric
acid as a by-product. I make the SO2 by dropping HCl onto sodium metabisulphite, its neater than burning and it can be done in glass (that SO2 smell
is something you'd prefer to avoid getting into everything).BromicAcid - 12-6-2011 at 16:47
Regarding metal salt catalysis... from a post I made in June 2005:
Quote:
IF... someone gets a good sulfur burner design that works. I found an interesting method to produce sulfuric acid that is simple and could easily be
made continuous. The premise is to bubble SO2 and oxygen into a collum filled with water (in the experiments they used 100 ml) with a small precentage
of manganese sulfate. The solutions can be made readily to concentrations of about 40% where the effectiveness of this procedure slacks off. Reading
though the article it looks fairly promising (but I cannot upload it because it is >1 M).
Recovery of Sulfur Dioxide as Dilute Sulfuric Acid
Raymond L. Copson, John W. Payne;
Ind. Eng. Chem.; 1933; 25(8); 909-916.
OK, I recall reading in a thread that a much respect member of this forum has tested this route to H2SO4 from H2SO3 with some success:
1. Saturate aqueous solution with SO2.
2. Treat with Cl2.
3. Cycle through Steps 1 and 2, ending at Step 1 to remove any residual Chlorine smell.
Chemistry:
Cl2 + H2O <---> HCl + HOCl
H2SO3 + HOCl --> HCl + H2SO4
Source: "Watts' Dictionary of Chemistry", Volume 2, Page 16, to quote the relevant section from Watts':
"Reactions.--1. HClOAq acts generally as an oxidiser; it easily parts with 0 while HClAq remains. Thus, As is rapidly oxidised with evolution of
light; P, S, Se, Br, I are converted to H3P04Aq, H2S04Aq, &c., even by dilute HClOAq; lower oxides or salts are converted into higher, e.g. SO2Aq
to H2SO4Aq, FeO to Fe203, As203Aq to As2O5Aq, FeS04Aq to Fe2(S04)3Aq, Fe2Cl6Aq, and Fe2O3, MnSO4Aq to MnO2; sulphides yield sulphates, c.g. H2SAq
gives" H2SO4,Aq and S; NH3 gives N,H2O,and NH4CLAq; HC1 forms H20 and Cl. The quantity of the acid expressed by the formula HClO oxidises the same
mass of an oxide"
Normally, just bubbling Chlorine through water is not very efficient, but in the presence of SO2, the reaction proceeds rapidly. Note, this process
consumes H2O (permitting the formation of a concentrated acid solution) and the volatile HCl is removable by heating, or by blowing it out with air
(or air and oxygen, see: http://www.sbioinformatics.com/design_thesis/Chlorine/Chlori... ).
I will defer any further comments and let the distinguished member comment if he so desires.
[Edited on 17-4-2012 by AJKOER]White Yeti - 17-4-2012 at 17:32
How about oxidising sulphite (obtained by bubbling SO2 through water) with an enzyme.... say sulphite oxidase? Or you could use an acidophile that
produces this enzyme.
Not sure if this is a viable method, but hey, no one has mentioned it before so I thought I might throw it out there. You'd have to find a way of
removing the acid so it doesn't kill the bacteria, not a simple task.weiming1998 - 17-4-2012 at 18:30
An alternative way that doesn't involve SO2 is on this thread: http://www.sciencemadness.org/talk/viewthread.php?tid=18963
Just react oxalic acid with MgSO4/FeSO4/CuSO4 in aqueous solution, and the insoluble oxalates should precipitate out, leaving you with a solution of
H2SO4.




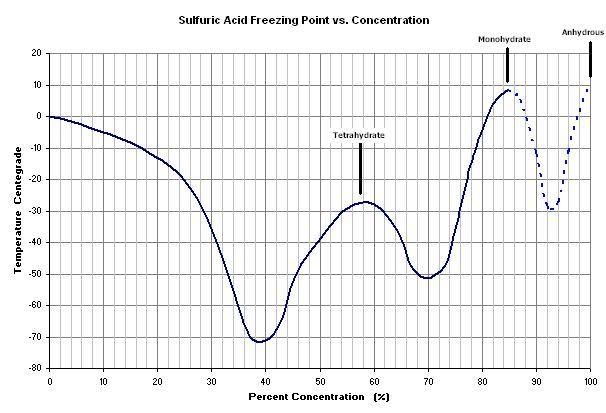

 .........which
.........which
