
The Mad Plater - 13-11-2016 at 15:32
Hello everyone, (this is my first post here on SM)
I'm currently working on developing a "high phosphorus" electroless nickel plating process for my personal use. (see note 1)
My main reference on this subject is "Electroless plating: fundamentals and applications" by Glenn O. Mallory and Juan B. Hajdu. (it's on Google, if
you want to take a look)
Other than that, I've dredged the available (free) publications, safety data sheets, and patents, for whatever tidbits I could find.
Based on the above, and several initial small-scale tests, I've come up with my own recipe. (see notes 2 and 3)
So far, I've had a mixed bag of results with this - everything works flawlessly up to the first bath replenishment, but then things tend to get
unhinged.
Here's my "homebrew" recipe for the initial bath makeup:
- 35g/L nickel(II) sulfate heptahydrate,
- 42g/L sodium hypophosphite monohydrate,
- 28.5g/L citric acid monohydrate,
- 60mg/L copper(II) sulfate pentahydrate (yes, that's miligrams per liter - it's a potent stabilizer),
- add NaOH to get pH up to ~4.5 (approximately around 15-16g/L)
Note: glass containers ONLY! (or at least glass-lined, ie. enamelled pots)
In a very unlikely stroke of luck, this actually works perfectly fine, and results in a deposit with the desired properties, as long as you keep the
pH, temperature, and reagent concentrations in the correct ranges, and discard the bath after the limiting level of metal turnover has been reached.
(see note 4)
So far, so good. Now, here's the kicker:
Look at the bottom of page 58 (that's chapter 2) of the abovementioned reference.
Paraphrasing, "for every mole of Ni+2, 3 moles of H+ are generated". Uh oh.
At this point, it would be a good idea to mention that I'm an engineer/machinist/mechanic, NOT a chemist. My knowledge of chemistry is, at
best, at a basic high school level - just enough to get me into trouble, but not nearly enough to sort out the resulting disaster.
Now, either there's a major discrepancy right there in the literature, or I'm misunderstanding some basic facts, because this doesn't jive
with the results of my testing. (no two guesses as to which of these causes is the more likely one)
Here's the deal:
I ran a bunch of small-scale tests (50mL bath), plating a very thin (0.01mm) piece of shim stock of known area (about 1/2 of recommended max bath
load) for a set time.
This resulted in approx. 20-25% of the reagents being depleted, which was verified as follows:
I miked the plated shim, and calculated the volume of the deposited alloy. From that, knowing the density, approximate alloy composition, as well as
the A.W. of nickel metal, I calculated the weight of the deposited nickel metal, and thus the # of moles.
Based on that, I calculated the amount of NiSO4 and hypo to add for replenishment - easy enough.
Obviously, the next logical step was to add 3x that # of moles of NaOH, to cancel out all that H+ that was generated, and get the pH
(roughly) back to the starting point.
And yes, the pH did in fact drop slightly during the plating operation, but it was still within the acceptable range at all times.
And thus we arrive at the root of the problem:
Adding the calculated amount of NaOH precipitated a major disaster, taking the pH all the way up to 11 (literally...), therefore causing the
("acidic") bath to rapidly decompose - precipitating the remaining nickel content as a fine mesh suspended powder, blackening the bath, with the end
result of rendering it completely useless. Good thing that was at a 0.05L scale...
Second test, same methodology, at the same scale - this time, I added slightly under 1/2 of the calculated amount of NaOH, and still the pH shot up to
over 6 - not nearly as bad, and no decomposition this time, but still WAY out of range.
Unfortunately, I failed to actually titrate the NaOH that time as well, as I was simultaneously experimenting with a "clever" way of preparing the
replenisment solutions - no, that of course failed miserably at what it was intended to achieve, but otherwise had essentially no significant effect
on the actual solution composition.
However, based on what I'm seeing, it seems as if there is approximately only 1 mole of H+ generated per mole of nickel deposited, which
can't possibly be right.
So, what actually happened to the remaining 2 moles of H+? Where did they go?
(see, told ya I'm no chemist!)
Any ideas, anyone?
Notes:
Note 1: No, I can't just send the parts off to a plating shop. Oh, how I wish I could. If only everything would be so easy and simple.
Reasons for this:
- I'm dealing with relatively infrequent occurences of unique one-off parts, or sometimes very small production runs (up to 5-10 parts), frequently
of unknown material composition ("mystery {metal}" - ie. the bane of every commercial plating shop;
- the parts are VERY valuable due to the amount of NRE and machining involved, we're talking high 3-figure prices per piece at the minimum;
- frequently there is NO room for failure - any plating screwup (ie. improper/no masking, too much coating thickness, etc.), and the parts are a
total loss;
- the customer (me) demands very high, consistent quality, as well as specific coating properties;
- the nearby plating shop (only 1 in the whole town) is ABSOLUTE AND UTTER GARBAGE, might as well toss the parts into the scrap bin before
getting them plated there, and it still saves time and money;
- I know exactly one halfway-decent plating shop (I use their services at my workplace), but it's very far away, they hate dealing with one-off
parts, and occasionally they will do a massive screwup, ie. recently they nickel-plated a batch of VERY expensive, high precision aluminium parts
which were clearly labelled "{color} anodize".
Note 2: I need a specific combination of properties - to name a few - NO porosity (amorphous structure), outstanding corrosion resistance,
excellent adhesion to substrate, decent microhardness and wear resistance, internal stresses compressive (or at worst, non-tensile), good visual
appearance (no discolorations, spotting, streaking, etc).
This invariably implies a high-phosphorus coating, as it's the only type which can potentially meet all these requirements.
I've looked far and wide, but to no avail. Couldn't find any available (non-secret) recipe that would satisfy the requirements.
It also doesn't help that the high-P process tends to suffer from low plating rates - and that means $$$ - so everyone in the industry wants to avoid
it if at all possible (medium-P plates much faster, and is thus the "default" choice), so there's a relative scarcity of information regarding it.
Note 3: It would all be very easy, if I wanted to do this on a large scale - industrial suppliers abound who sell such processes, based on
their proprietary concoctions.
Unfortunately, they won't even pretend to be interested in selling anything, unless you're dealing with quantities in the 100s of liters range.
Also, they will normally refuse to sell anything to individuals, even if you actually wanted a "large" quantity for whatever reason.
Note 4: The metal turnover life of my "concoction" is pitifully low, effectively only 1 metal turnover - but I consider this as being perfectly
acceptable, because:
- this is being done on a fairly small scale, on the order of 5-10l of bath utilization per year at most,
- the bath replenishment calculations essentially involve educated, approximated guesswork, which will tend to diverge badly from reality with each
subsequent metal turnover (the errors are cumulative),
- the "spent" bath is still less valuable than the amount of my time involved in the remainder of the plating process, to say nothing of the surface
prep,
- in any case, both the remaining value of the bath AND the value of my time spent on the plating process is totally peanuts compared to the value of
the parts being plated, and also the devastating costs potentially incurred by a plating screwup.
elementcollector1 - 13-11-2016 at 16:24
I actually had to do something like this for a university research project last year. If I recall correctly, the plated material would give off
hydrogen gas while being immersed in solution. This may be why your 3 moles of NaOH were 2 moles too many - 2 of the H+ ions were lost to form
H2.
m1tanker78 - 13-11-2016 at 17:40
elementcollector1 is right, electroless nickel plating releases hydrogen gas (small bubbles form on the part when it's submerged in the bath).
You probably already know that NaOH is crazy hygroscopic which is why an exact mass is avoided. How are you determining the pH when you add NaOH? How
do you dispose of the spent batches of plating solution?
WGTR - 13-11-2016 at 18:24
Among the reasons why I hate electroless nickel:
1. It's hard to replenish, as you've discovered. Eventually the bath has to be dumped, and remade. I have used products from Caswell with moderate
success.
2. There are difficult to control process variables. Temperature and stabilizer content are a couple. If the part has too much or too little surface
area for the volume of plating bath, the stabilizer can go out of tolerance, causing the bath to decompose or not plate at all. I can't explain why
this is. Slight temperature variations can cause significantly different results from part to part. It's better to have a large plating bath to
allow better temperature control. It may be necessary to "dummy" the bath with a steel plate to keep the inhibitors in spec if plating a small part.
When the bath decomposed, was there a lot of simultaneous out-gassing?
Adding NaOH solution to the bath most likely caused localized increases in pH during addition. Once the bath begins to decompose a little, then the
decomposition accelerates.
Developing a custom plating bath can take a lot of time and money. I developed a specialized bath for a particular part, and it took about a year to
work out the bugs and complete the testing. Now it works perfectly every time. Have fun.
The Mad Plater - 13-11-2016 at 21:20
Duh! Of course I never even considered the offgassing.
Yes, I fully expected that it would be hydrogen - did read about that someplace or another.
But I totally failed to consider where it was coming from!
Don't know what was I thinking.
Well, in any case, thanks for pointing that out.
WGTR:
Trust me, if I could get a prepackaged "kit" like that (even if just the chemistry, as I already have the other equipment), I'd do so in a heartbeat.
One of the most important things you learn as an engineer (in practice, not through education), is "don't reinvent the wheel". Off-the-shelf hardware
is the way to go, whenever possible.
Well, there are 2 main problems here:
1. The Caswell EN kit is medium-P, which I have no need for,
2. Their plating kits don't seem to be available locally (but I didn't really bother checking up on this, because see point 1 above).
The temperature gave me the least trouble, despite the fact that I was running this in a tiny 100mL beaker on a gas-fired stove burner (on a
mesh, obviously).
Next on the shopping list is an electric hotplate. If the built-in thermostat turns out to be inadequate, I'll just buy a "cheap" PID temperature
controller and hook it up. Piece of cake.
If that turns out to be inadequate, plan B is to buy a cheap deep frier, fill it with water instead of oil (the oil would work fine, but it's an awful
mess), and put the beaker with the bath into that. The extra mass of water would further decouple the beaker from the heating element, and by
regulating the outer bath temperature as the primary setpoint, the temperature swings (as seen by the plating bath) are minimized.
The bath load is a minor issue for me, since I don't anticipate the need to deal with heavily masked parts (we're talking maybe 10-20% masked area at
the very most), and the parts tend to have reasonable shapes, ie. "short and stout", not very long and thin - or even worse, long, thin, and bent into
a pretzel.
All that having been said, I can keep the bath load reasonably consistent for the various jobs.
As for the madness you've observed - part of the problem is, some substances are VERY potent stabilizers, for example, IIRC, a few ppm of lead will
stop the reaction dead in its tracks.
When that test bath decomposed, it first turned merely hazy, and then over the next few minutes started turning completely black. I sure as hell
didn't want to stick around to see what happens next (and possibly trash my beaker with insoluble nickel crap), so I got rid of it at that point.
As for that localized decomposition, well, I got that covered. Found out the hard way during initial makeup.
Now I add the reagents in a specific order, allowing the previous one to dissolve fully before adding the next.
Also, stir stir stir. And then stir some more. And then even more, for good measure.
m1tanker78: I weigh the NaOH straight out of the bottle for the approximate "initial" addition, then I use a 2M solution in a syringe (remember, we're
still talking small scale) to fine tune the pH.
For measuring the pH I use narrow-range pH papers. Not ideal, I know, but a pH meter is out of my price range, and IMO too cumbersome (calibration) to
use on a once-in-a-blue-moon basis.
The papers are cheap, fast and easy to use, and they do the job OK.
As for disposal, well - "the solution to pollution is dilution".
Our (municipal) sewage goes through a waste treatment plant, and we're paying through the nose for that.
They can handle this.
And before anyone complains of "OMG HAX! ur recking da panet earf!" - well, the trillions of liters of raw untreated sewage being discharged annually
straight into the waterways have done quite a number on it already.
(Which, incidentally, helps explain why the water in our rivers and lakes tends to look like raw sewage - because it contains so much of it!)
Metacelsus - 14-11-2016 at 05:23
In any case, the citrate should act to buffer the system, so I wouldn't worry too much about large pH fluctuations.
wg48 - 14-11-2016 at 05:41
Welcome to the forum Mad Plater.
Ok as evidenced by your comment "educated, approximated guesswork" you do understand the potenial difficulties of equating total plated nickel to
changes.
The commonly quoted ratio between H+ to Ni of 3:1 is only approximate and includes the hydrogen generation. Thats the extra mole of H+.
Edit: I was in a rush with my first answer. I have deleted the gobbledegook now. The following is a better version
From: http://nvlpubs.nist.gov/nistpubs/jres/39/jresv39n5p385_A1b.p...
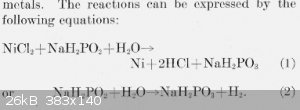
So you have to counter the acidity (or alkilinity) when NaH2PO2 changes to NaH2PO3. Its not as simple as equating the H ions.
I have tried electroless nickel plating without any successes. I was trying to plate plastic using silver and or tin as the sensitiser but had no
success.
I will try your formulation as I have all the ingredients. What is the recommended plating temperature and what plating rate should I expect?
[Edited on 14-11-2016 by wg48]
The Mad Plater - 14-11-2016 at 08:17
The citrate does in fact buffer the pH fairly reasonably - but, AFAIK, the optimal range to get a good coating is quite narrow.
In this case, "large fluctuations" would be anything over +/- 0.5; too acidic and it stops plating, whereas too basic messes up the alloy composition,
and also results in poor adhesion.
The plating rate is strongly pH-dependent, so closely controlling the pH is the key to getting any kind of repeatability out of the process.
Fun fact - the first recipe I tried (pulled unchanged straight from some publication) was based on sodium acetate as the buffer, instead of citrate.
That particular test came out OK - although it was a medium-P formulation, so not really what I needed - but the smell was rather disagreeable... at a
50mL scale.
Needless to say, that bottle of sodium acetate was promptly relegated to the rear of the "infrequently used junk" cabinet.
wg48: that ~1:1 ratio is certainly consistent with the observations so far.
The guesswork goes much deeper than that.
Right now, I'm testing this on very simple test pieces, the plated area of which can be determined reasonably accurately, say to +/- 5%.
OTOH, the actual parts tend to have rather complicated shapes, which makes accurately estimating the area quite a difficult and laborious process.
So I'm guessing the amount of nickel deposited, based on the thickness measurement (very accurate) times the estimated area, plus a bunch of
other assumptions.
Other interesting things:
Still can't figure out how (or if it's even possible) to make a single-part replenishment solution. Specifically, how to make the solution suitably
basic, for bath pH control, and still be able to keep the metal sulfates in solution.
I managed to completely obviate the need for conc. HCl in this process - the pickling/activation step works perfectly fine with citric acid at an
elevated temperature, just takes a bit longer (a few minutes).
Which is great, because conc. "strong" (in the corrosive sense) acids is where I draw the line. I refuse to use them, unless there are really no other
effective, safer alternatives.
I've had the experience of working with the relentlessly corrosive hydrofluoric acid before (yikes!), as a premixed component (~20% IIRC) of stainless
pickling gel - I don't think there is any alternative "safe and effective" way of doing that, sadly.
Well then, the plan for the next test seems to be roughly as follows:
- plate a shim, determine # of moles of plated nickel, etc, yadda yadda yadda,
- add NiSO4, hypo and CuSO4 to replenish the spent reagents,
- carefully titrate with NaOH, determine the required molar ratio,
- plate some more shims made of tinfoil, test them for non-magnetism (amorphous nickel alloy structure = high-P), and again measure the amount of
plated nickel,
- repeat the last 3 steps until the bath is spent (note: determine the actual endpoint of bath life)
Ack - that'll easily take all day!
Then there are more unanswered questions which require extensive testing, ie.:
- the alkaline precleaning step with trisodium phosphate,
- good masking materials and methods,
- adherently plating EN on aluminium (via the zincating process).
And next... the remaining processes to develop, in that order:
- manganese phosphating,
- zinc phosphating,
- anodizing,
- stainless electropolishing.
AFAIK, the last 2 require sulfuric acid - luckily, relatively dilute.
I've run some preliminary tests on electropolishing without sulfuric acid a few months ago, and got nothing. All the available recipes seem to include
it.
I'm fairly convinced that it's a key ingredient, which can't be replaced with anything milder, at least not without reformulating the whole process.