
DrDevice - 7-7-2018 at 01:14
I have recently been experimenting with the preparation of the azlactones (or oxazolones) from aromatic aldehydes. They are relatively straightforward
to synthesize, but with the drawback that the necessary acetic anhydride can be difficult to obtain.
Looking at the azlactone synthesis, it can be seen that both acetic anhydride and sodium acetate are required. As I have acetyl chloride and sodium
acetate, I decided to try preparing the acetic anhydride in situ with the action of acetyl chloride on an excess of sodium acetate, leaving the sodium
chloride by-product in place for the rest of the reaction.
The most relevant published paper to this procedure is "Azlactone of alpha-benzoylamino-beta-(3,4-dimethoxyphenyl)-acrylic acid", Org Syn Coll V2 p55
(1943), doi 10.15227/orgsyn.013.0008, but there are quite a large of number of similar published preparations. For example, there are papers on the
process on vanillin and substituted vanillin, which I have also performed successfully.
Procedure
36.2g (0.44 mol) of anhydrous sodium acetate was placed in a 2-necked 250ml flask set up with a dropping funnel, a reflux condenser and magnetic stir
bar and placed in an oil bath that was initially at room temperature. The top of the reflux condenser was capped with a calcium chloride drying tube.
24.8g (0.32 mol) of acetyl chloride was placed in the dropping funnel and allowed to drip at a rate of about 1 drop per second into the slowly
stirring sodium acetate powder. A thick paste forms over several minutes, and the stir bar occasionally "jams" during the process.
On completion of the addition of the acetyl chloride, the oil bath temperature is set to 60C and the mixture is heated for 30 minutes with stirring.
Only a small amount of the solid sodium acetate is mixing at this point, most is caked on the wall of the flask.
After 30 minutes, the flask is opened and the caked solid is pushed down to the bottom of the flask as much as possible from the flask walls.
12.4g (.069 mol) of Hippuric acid and 12.42g (0.063 mol) of 3,4,5-trimethoxybenzaldehyde is added, the dropping funnel and reflux condenser is
removed, but the calcium chloride tube is re-connected in place of the reflux condenser. The oil bath temperature is set to 90C.
The thick pasty mixture soon develops a yellow tinge, and magnetic stirring becomes increasingly difficult and the stir bar can stall, but the
reaction continues nonetheless.
After 90 minutes, the reaction is complete, as indicated by TLC (1:3 EtOAc:hexane) showing elimination of all the 3,4,5-TMBA. The mixture is now a
vivid yellow-orange colour. It is allowed to cool to room temperature.
On cooling, the mixture in the flask is quite solid, Water is added, and some effort must be used to break the product into lumps small enough to
remove from the flask.
The reaction products are placed in a beaker with cold water and stirred until all large pieces are broken up. Clearly visible is the yellow-orange
azlactone suspended in the water. The azlactone is filtered and washed twice with water.
Recrystalization of the product can be done using boiling EtOH. Note that a very large quantity or EtOH is required - for the quantities used in this
procedure, 1.2l was required. Commercial grade "Methylated Spirits" was used with no apparent ill-effect.
The azlactone was allowed to precipitate out overnight from the cooling EtOH. The next day, the very fine bright yellow-orange powder (no real crystal
form was noted) was separated by filtering and dried gently on a hot plate.
Yield obtained : 16.3g, MW of the azlactone is 339.34 so a yield of 76% from the original aldehyde. MP = 159 - 162C. No literature source to compare
this against has been located.
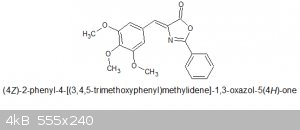
Discussion
The ratios I was using in reactants for aldehyde:hippuric acid:sodium acetate:acetyl chloride were 1:1.1:7:5, in an attempt to get some solvation
happening. In future, I will try to add some other solvent for example cyclohexane to help in the mixing process. But as can be seen from the yield,
it was still yielding at about the same % as the procedures using acetic anhydride.
TLC was performed at 15 minutes, and 1 hour. During the experiment, I used a 1:1 mix of EtOAc and hexane, and the Rf was quite high - 0.86 for the
aldehyde, and 0.96 for the azlactone, but the reactant and product were still quite distinct. After 1 hour, only a very faint trace of the aldehyde
could be seen. On the TLC for the purified product, and the different eluent ratios, the Rf for the aldhehyde was 0.47 and 0.58 for the azlactone.
Only a single (bright yellow!) spot was seen on the TLC for the product.
Without MP data from the literature, I guess it is a bit hard to definitively claim that I have ended up with what I hope. All I can say is that the
colour is right, and the measured MP is in the range of other methoxy-substituted azlactones.
The recrystlization was a pain. Other azlactone procedures suggest other solvents such as acetic acid or ethyl acetate (or even benzene). I will be
trying with acetic acid with a small quantity soon.
One of the most interesting things of the azlactone reaction is that the result is a C-C double bond formation. Further processes on the azlactone can
open the ring and lead to amino acids and phenylpyruvic acids.
Loptr - 7-7-2018 at 05:16
What would lead to the amino acid? First glance makes me think you would easily end up with an alpha-hydroxy acid.
DrDevice - 7-7-2018 at 06:09
Amino acid synthesis was one of the first results obtained from azlactones by Erlenmeyer back in 1893.
The literature describes methods for "reduction and hydrolysis". Organic Reactions, Chapter 5 "Azlactones" (Carter) gives an overview. Org Syn Coll
Vol 2 p489 shows a method for reduction of the azlactone derived from benzaldehyde being converted to phenyalanine.
These are all based on sodium or sodium amalgam, hydroiodic acid/phosphorus, or catalytic hydrogenation, none of which are terribly easily accessible
to the home chemist. However, the NiCl2/Zn method (eg "Studies of Reduction of Various Organic Compounds with the Nickel(II) Chloride - Zinc System",
(Nose, Kudo, 1990) is more accessible...will try that first.