Heptylene
Hazard to Others

Posts: 319
Registered: 22-10-2016
Member Is Offline
Mood: No Mood
|
|
Sizing Rashig rings for Hempel column
I'm want to get a new column for some upcoming distillations. The column will also double as a packed bed absorption tower for my Ostwald reactor. The
column will likely be 700 mm (36 mm OD), though there are 500, 800 and 1000 mm models available. I've never used packed columns before so deciding on
which size Raschig rings to use is tricky.
I can get the following sizes for rings (OD x length in mm):
10x10, 8x8, 6x6, 5x5. The wall thickness is unknown, likely 1 mm.
Apparently smaller rings give better separation but more product gets stuck in the column at the end of the batch.
I plan on distilling mainly nitric acid (hopefully 68% from <10 % in one distillation step) and ethanol (from cheap wine or mash for use as a
solvent). Possibly gasoline (?) for fun too.
Does anyone have useful info to share on this?
|
|
|
Praxichys
International Hazard
Posts: 1063
Registered: 31-7-2013
Location: Detroit, Michigan, USA
Member Is Offline
Mood: Coprecipitated
|
|
A 700mm Hempel column is total overkill for most of those things. The problem with a column that big is that the reflux ratio is astronomical.
Although the separation efficiency is high, your throughput for a given energy input will be very low and you'll likely need to insulate it to prevent
flooding assuming its ID is around 20mm. Your distillations are going to take an unnecessarily long time, and as you mentioned it will retain a lot of
product and be a pain in the ass to wash.
You will find that the nitric acid and ethanol distillations can be done in a couple of passes with a simple distillation kit in far less time than a
single pass through pretty much any packed column. In my ethanol setup (which is just a pressure cooker with some copper tube attached to it) I can
pull 70% EtOH off on the first pass with zero column or packing from a 14% mash using turbo yeast and cane sugar. After that, you can use potassium
carbonate to float the ethanol off the remaining water, then dry using sieves and redistill if you absolutely have to get rid of all traces of salts.
As for nitric acid, I usually use the H2SO4/KNO3 route but you should get mostly water until the pot hits 68% and then hover at the azeotrope
thereafter, even with simple distillation.
Keep in mind that using glass as a first pass for anything you need a lot of gets expensive quickly. Even with a 1 liter flask (which can safely hold
about 700mL) you're barely going to get 100mL of 68% nitric starting from 10%, and much less with any column holdup. Same with ethanol... if you need
a liter of it, you'll have to run 11 batches at the 1L scale from 14%. It's much easier to bodge some stuff together to rip it most of the way before
going to the glass.
Anyway, I feel like I'm rambling now. That said, I have a 500mm 24/40 Hempel (roughly 20mm ID) and I have packed mine with 6x6 rings (chopped 6mm
flint glass tube, actually) which works well and "feels" like a good size, satisfactorily creating petroleum ethers with custom boiling ranges,
separating benzene from pyrolysis oil, etc.
|
|
|
Sulaiman
International Hazard
Posts: 3558
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Offline
|
|
As above, a 35mm o.d. x 700mm tall packed glass column used to distill nitic acid or ethanol
will need thermal insulation to get any product through it.
Although the column would only dissipate about 70W for ethanol and 100W for nitric acid,
the bottom of the column will almost certainly flood.
As you are aiming for a relatively large number of theoretical plates,
temperature stability within the column will be very important
- a small change in external air velocity will completely destabilise the column.
I like rockwool pipe insulation like this https://www.paroc.com/products/technical-insulations/ti-pipe...
(a simple to use heat loss calculator: https://calculus.paroc.com/paroc-calculus/#/ )
Such a tall column will not fit in a fume cupboard ... for reference,
here is my first attempt at a 700mm (2x 350mm, 550mm actual packing height) packed column vs ethanol ...

(Two product condensers was overkill - used just because I could)
This setup was a failure as during winter the reflux condenser was so efficient that I got no product,
I should have insulated the reflux condenser also 
I now use a different still head adapter that allows the product condenser to be vertical, parallel with the column,
a great space saver and only one (substantial) vertical rod for the clamps.
In my opinion, a 700mm column should only be used if you want very fine separations,
such as collecting the ethyl acetate produced during fermentation, separated from ethanol.
___________________________________________________
Here is some data that I collected on ethanol fermentation;
WHAT TO EXPECT:
Acetone 56.5C (134F)
Methanol (wood alcohol) 64C (147F)
Ethyl acetate 77.1C (171F)
Ethanol 78C (172F)
2-Propanol (rubbing alcohol) 82C (180F)
1-Propanol 97C (207F)
Water 100C (212F)
Butanol 116C (241F)
Amyl alcohol 137.8C (280F)
Furfural 161C (322F)
http://homedistiller.org/forum/download/file.php?id=2379&...
Typical malt distillery analyses
(in grams per 100 litre absolute alcohol)
Acetaldehyde 5.4 7.0
Ethyl acetate 26.3 33.2
Diacetyl 2.1 2.8
Methanol 4.8 9.0
Propanol 40.3 37.6
Isobutanol 80.1 85.6
o.a. Amyl alcohol 44.2 53.1
Iso-amyl alcohol 138.9 170.8
Total higher alcohols 303.5 347.1
Ethyl acetate 3.7 5.3
Ethyl octanoate 1.8 2.7
Furfural 3.6 4.8
Ethyl decanoate 6.0 8.9
b-Phenethyl alcohol 7.2 8.7
Ethyl myristate 0.9 1.2
Ethyl palmitate 2.8 3.3
Ethyl palmitoleate 1.6 2.1
Phenols 1.0 4.5
___________________________________________________
Honestly, I have not got the 700mm column working as I wish yet - so many things to control.
So, unless you want a MAJOR project to complete,
I suggest that you go with a c350mm column and not use a reflux condenser with a partial takeoff system.
Anything above 5 theoretical plates gets really complicated.
___________________________________________________________
@Praxichys: 14% to 70% for a simple distillation of ethanol:water ... seems optimistic.
[Edited on 22-10-2019 by Sulaiman]
CAUTION : Hobby Chemist, not Professional or even Amateur
|
|
|
markx
National Hazard
Posts: 645
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
I would suggest to not try and create a "fit all situations" setup....as in suitable for both nitric acid and alcohol distillations in a fine
fractionation mode. These are quite different animals that likely do not bode well in the same cage 
In all probability you will end up with a system that does not do well in either situations and drags unneccessary complications along construction
and operation stages. In my opinion it makes sense to keep the systems separated.
Keep the nitric system as simple and small as reasonably possible. Likely you do not need gallons of it being processed at a time. There will be
mishaps and leaks along the way, so dealing with a smaller system in way more healthy. Also the cost, as mentioned before, will skyrocket with
expanding dimensions. So will the grief if one manages to break something.
The alcohol system can be more sizeable and constructed from more reasonable materials. Stainless and copper are the best choices for that project.
Plumbing supply stores carry suitable fittings to construct a very decent apparatus with minimal machining, welding and soldering required. I would
steer away from using glass as the main element in a sizeable system. It is awkward, fragile and expensive. One can not really modify or rebuild it if
there is a need for it....
As for getting a 14% mash up to 70% in a single simple distillation run....totally doable, but the percentage will drop heavily as the mash depletes.
If one has the patience to let it drip out on a real slow heat (reflux ratio inside the system increases when heat input is lowered compared to losses
through the system) then a good sized fraction of 70% can be obtained. Still even a mediocre column will do wonders in terms of proof, purity and
output compared to the most carefully executed simple distillation run.
Columns are really not too complicated beasts, as long as one has left room to regulate the heat input, cooling capability and packing type/density
then it is not too hard to get them to operate properly with an excess of even 10 theoretical plates. But it is usually impossible to get them just
right with the first try, even with previous experience, so leave some room for modifications.
[Edited on 22-10-2019 by markx]
Exact science is a figment of imagination.......
|
|
|
Sulaiman
International Hazard
Posts: 3558
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Offline
|
|
Quote: Originally posted by markx  | | ... As for getting a 14% mash up to 70% in a single simple distillation run....totally doable, but the percentage will drop heavily as the mash
depletes. If one has the patience to let it drip out on a real slow heat (reflux ratio inside the system increases when heat input is lowered compared
to losses through the system) then a good sized fraction of 70% can be obtained. Still even a mediocre column will do wonders in terms of proof,
purity and output compared to the most carefully executed simple distillation run... |
I agree with the last sentence, but ...
a) if you are refluxing then you can't call it a simple distillation
b) I'd like to know what yield of 70% ABV you can get from a 14% ABV mash without a reflux column.
... somewhere between negligible and zero based on my experience.
(but if you can demostrate otherwise I'd appreciate it)
EDIT: this may help clarify my point of view
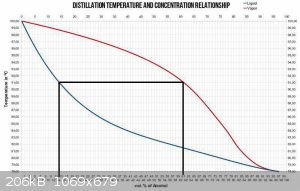
And as you are aware, both of the vertical lines move to the left as product is removed.
[Edited on 22-10-2019 by Sulaiman]
CAUTION : Hobby Chemist, not Professional or even Amateur
|
|
|
Praxichys
International Hazard
Posts: 1063
Registered: 31-7-2013
Location: Detroit, Michigan, USA
Member Is Offline
Mood: Coprecipitated
|
|
I don't meant to hijack the thread but here are the details of my apparatus/process:
The distillate measures around 70% using a crappy eBay hydrometer. The still has a 5 gallon capacity and it starts coming off around 85%, and I cut it
off before 40% or the yeast starts burning and giving the product a nasty smell. I get around 4.5 liters of distillate.
The still is a Presto 5 gallon pressure cooker with the gauge removed. The hole (center of the lid) is drilled out to 1/2". A stainless 1" NPT floor
flange is affixed over said hole using 4 stainless bolts and silicone as a gasket. Threaded into this is a 1" copper pipe extending vertically about 2
feet, intended to be a packed column but never actually packed with anything (yet). An elbow at the top of this reduces to a 3/8" compression fitting
and a length of 3/8" copper tube with a PVC water jacket with hose barbs for tap water flow.
This is placed on a gas stove (ca. 12,000 BTU) cranked to max for the whole run. The distillate is captured as a steady stream and is complete in just
under two hours, until a sample taken into a graduated cylinder falls below around 40% by hydrometer.
It is currently in storage (I just moved) but I have dug up some old (and kind of shameful - my kitchen was a wreck at the time and Google Drive won't
let me rotate them) photos:
https://drive.google.com/open?id=0B_16NzkNoL75UlR3T1dFN3I1Ml...
https://drive.google.com/open?id=0B_16NzkNoL75N0tFZnllOWpqaU...
https://drive.google.com/open?id=0B_16NzkNoL75RlZ2dVQ0M3RBek...
My washes use "Turbo Pure 48" with sugar stoicheometrically added to ferment to 20%. I let it run for around 4 days but without external heating or
cooling so the washes probably end somewhere closer to 18%. It still tastes sweet suggesting an incomplete fermentation even when activity is
basically zero. I haven't bothered actually checking since this is always "quick and dirty" and I don't really care to control the temperature much
since the raw materials are so cheap, really only removing the insulation if the temp climbs past about 82°F. I use a 5 gallon bucket wrapped in
carpet with a 2L bottle serving as a bubbler to monitor activity. No particular attention is paid to sterility, with washes started using only hot tap
water and granulated sugar weighed with a bathroom scale.
https://drive.google.com/open?id=0B_16NzkNoL75V0dnQk9sSm5PeX...
I will usually "salt out" the first-run distillate with about 1.5 kg of technical K2CO3, pouring off the supernatant alcohol and allowing it to sit
over a little more K2CO3 for a week or so to hydrolyze any errant esters and to polymerize any ketones. This also gets rid of the yeast-y smell. The
carbonate is recovered by leaving the aqueous layer in a glass pan outside in the sun until any residual alcohol is gone, then baking in the oven
until dry. It will take on a sandy brown color from decomposing carboxylic acid salts but can be burnt white by running the oven through a cleaning
cycle with the K2CO3 inside.
The ethanol is then redistilled (this time from glass) to remove any dissolved salts, with a heads/tails discarded to assure purity, leaving on
average around 2.5-3L of EtOH for a total cost of about $12 and a few hours of work over about 2 weeks.
Yields:
Theoretical yield from sugar: 3.8L (19 liters @ 20% ABV)
From sugar, first distillation: approx. 83% (4.5L @ 70% ABV) - if 100% recovered, suggests a 16.5% wash
First distillation to final product (2.5-3L): 79%-95%
Total yield from sugar: 66%-79%
I'm pretty sure my "column" is way overloaded by the massive heat input combined with being in the exhaust plume of the gas stove and probably has
nearly zero reflux. I think what might be happening here is that residual sugar plus the salts used as yeast nutrients in the turbo yeast packets are
acting as entrainers to sequester more water than would otherwise distill from a pure water/ethanol mixture, if I had to guess.
[Edited on 22-10-2019 by Praxichys]
|
|
|
Sulaiman
International Hazard
Posts: 3558
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Offline
|
|
When I'm reunited with my chemistry stuff I'll try some potassium carbonate
... my supposed-to-be 20% or 22% also come out at around 18% and after a simple distillation, keeping the heads,
it smells and apparently tastes awful (I have a designated test taster).
Typical 55% ABV after first distillation ... 0.3l to 2l scale in glass.
I try to keep all of the fractions as I'm interested only in the separations / distillation processes.
At the end of my distillations I mix all of the fractions (minus water) back together as stock for my next distillation experiments.
Due to restrictions of my religion I can't enjoy the product, or even allow others to enjoy it
[Edited on 22-10-2019 by Sulaiman]
CAUTION : Hobby Chemist, not Professional or even Amateur
|
|
|
Heptylene
Hazard to Others
Posts: 319
Registered: 22-10-2016
Member Is Offline
Mood: No Mood
|
|
Thank you all for your advice and experience. Any thread hijacking was welcome Praxichys! Clearly a 700 mm column is not as good a choice as I
thought. I'll probably end up getting a 500 mm/30 mm ID instead. This should suffice for most things. I guess given the smaller column diameter
Raschig rings of 6 mm are the best suited now.
I intend to use my column with a total condenser and liquid reflux divider, so I won't need to carefully regulate the condenser temperature.
Also regarding the limited batch size when using a glass boiling flask: There are ground glass to NPT threaded adapters on the market.
I think using a metal boiling pot and a glass column could work. Or even just a glass condenser for the first separation pass. This would save time
and money on making a custom copper condenser when a liebig condenser is basically the same thing, and most of us already have a liebig.
|
|
|
Sulaiman
International Hazard
Posts: 3558
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Offline
|
|
If you have a large pot then I assume that there will be a lot of heat input,
which needs to be removed by the reflux condenser (and some via the column),
so size your reflux condenser to match the heat input,
e.g. not a Leibig, West or Allihn.
and definitely not a Davies type that would blow out your product.
Something like a double surface or Graham or Dimroth would be more suitable.
Using definitions found here: https://en.wikipedia.org/wiki/Condenser_(laboratory)
Your ultimate throughput should be limitited only by the column ... not flooding.
For reference; my column is c25mm i.d. and using 6mm glass spheres ve ethanol:water
this condenser out-performs my column https://www.ebay.co.uk/itm/24-29-Jionts-Glass-Reflux-Graham-... (thanks to my SM SS ... j)
whereas a 250mm Leibig is too small.
OTOH
you do not want to cool the refluxed liquid more than necessary to condense it
or you will be dropping liquid that is colder than the top theoretical plate, onto the top of the column.
Complicated innit ?
[Edited on 23-10-2019 by Sulaiman]
CAUTION : Hobby Chemist, not Professional or even Amateur
|
|
|
markx
National Hazard
Posts: 645
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
| Quote: Originally posted by Sulaiman |
I agree with the last sentence, but ...
a) if you are refluxing then you can't call it a simple distillation
b) I'd like to know what yield of 70% ABV you can get from a 14% ABV mash without a reflux column.
... somewhere between negligible and zero based on my experience.
(but if you can demostrate otherwise I'd appreciate it)
EDIT: this may help clarify my point of view
And as you are aware, both of the vertical lines move to the left as product is removed.
[Edited on 22-10-2019 by Sulaiman] |
The point is that "simple distillation" setups rarely are truly simple in practice. So the phase diagram tends to develop at least a "vague"
representation of another separation step even in a pot still.
There shall inevitably be naturally occurring reflux activity on the walls of the boiler and tubes leading to the condenser. The fact that one does
not have a column in the system will not remove the effect totally. One can not control the ratio, but as heat input is decreased the uncontrolled
natural reflux ratio increases somewhat, hence you can conjure higher proof product from a pot still when working slow and careful.
As for the yields, it depends on the particular apparatus (glass vs. metal, insulation, size, the part leading to condenser etc.), but negligible
would be about right 
I for one would not bother to try and get a reasonable portion of a 14% mash up to 70% using a simple distillation setup. It can be done as I said,
but the time and effort invested is better spent on designing a fractionation column and learning to operate it. All the additional trouble of drying
with salts, then redistilling to get rid of the residues, filtering, etc can be avoided by a proper reflux system.
Flowing reflux that is somewhat subcooled back into the column is not really a catasthrophic event and for practical purposes it really can not be
avoided to be honest....all that it does is increace the energy demand somewhat (one is drawing out more heat than is needed for just condensation to
occur). It also shifts the last plate lower into the column and compresses previous ones. Not a tragedy....
I've mentioned this earlier: columns are mostly selfregulating systems when operated under acceptable conditions. Trying to measure, control, evaluate
and regulate every aspect of their operation is going to drive one mad and in reality accomplish nothing. Just let it operate and lean back....it will
find it's own sweet spot
[Edited on 23-10-2019 by markx]
Exact science is a figment of imagination.......
|
|
|
|









