Boffis
International Hazard

Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Preparation of cinnamalacetone
Has anyone on this board ever prepared cinnamyl acetone? I am trying to prepare this compound in the same way as benzal acetone but replacing the
benzaldehyde with a molar equivalence of cinnamaldehyde.
The method was as follows:
5gm cinnamaldehyde were mixed with 10ml of acetone (an excess) and 1ml of 10% sodium hydroxide solution and the mixture stirred at about 20C for 30
minutes and then left to stand for 2 hours at 25-27C with occasional stirring or shaking. The mixture was then neutralised with a little HCl and
diluted with 10ml of water. On oily yellow liquid with some curdy solid formed but it did not solidify completely so can't be filter off. The smell of
cinnamaldehyde is now slight but the melting point of pure cinnamalacetone is only about 62 C so impurities could easily drop the melting point to
room temperature. Any suggestions? ether or ethyl acetate extraction possibly?
Can cinnamaldehyde form a dicinnamal-acetone analogous to dibenzalacetone? Has anyone seen a general procedure for the preparation of symmetrical
di-substituted acetone. I am also looking at other possible derivatives such as divanilal- and disalicylal- acetones
Also, according to some of the more general reports I have read suggest that sometimes a hydroxy ketone first forms and that this then dehydrate on
further heating but heat also generates more side reactions and a darker product (from my own experience). Is there a way to judge which which
aromatic aldehydes will react directly to the unsaturated ketone?
By contrast a parallel experiment using 5g of 4-dimethylaminobenzaldehyde plus 10ml of 95% ethanol (to create a stable homogenous solution) after 3
hours virtually solidified into a crystalline red mass that filtered and washed easily after neutralisation. The crystals are a bit sticky but I will
recrystallise them tomorrow. The filtrate was cloudy so I tried extracting it with ether but very little was recovered, ether does not appear to be a
good solvent for this compound. One paper I found suggests ethyl acetate extraction (Synthesis 2008; No22, p3675).
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
Note 2 here has a prep of dicinnamalacetone based off of a 1885 Chem. Ber. paper. Having gone that route before of digging up old work and
translating, their modern version is guaranteed to be more detailed. The reaction is extremely rapid.
Adding the link would help: http://www.orgsyn.org/Content/pdfs/procedures/cv7p0059.pdf
As far as pure cinnamalacetone, I imagine a gross excess of acetone will result in the monosubstituted product. The solubility in water should be
extremely low. Have you tried triturating it with more ice water to reduce the effects of it being in 50% acetone.
[Edited on 26-10-2019 by UC235]
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
UC235 many thanks for that link. This will be very helpful for the disubstituted acetones I am goin to try next.
The oil in my two batches of cinnamalacetone has solidified into beautiful yellow crystalline masses (see photos) and has now been filtered off and is
drying. I did a second run because I thought I had screw up on the first one but in reality it was just a lack of patience that was the problem!

The left-hand photo is the solidified oil from the first batch while the right-hand photo is the double sized second batch which is made of masses of
needle shaped crystals

The 4-dimethylaminobenzalacetone batch was also successful yielding 5.57g of crude brick red crystals from 5.01g of dimethylaminobenzaldehyde.
Just need to see how these compounds can be recrystallised now.
[Edited on 26-10-2019 by Boffis]
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Another question! In the link posted by UC235 above the description gives an amount of NaOH to be used. If I were to use a phenolic aldehyde such as
vanillin should I increase the amount of NaOH proportionally (in Molar terms)?
The reason I ask is I tried the reaction with vanillin and it appeared to be working but with only a limit amount of vivid yellow ppt. Thinking that
most of the product was probably insolution as the sodium salt I added just enough acetic acid at the end to neutralise the NaOH use. All the
precipitate dissolved!! I have struggled my balls off to recovery something but to no avail. I imediately added some NaOH but no ppt formed.
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
Vanillylideneacetone is an undergraduate prep. I have done it several times. There are also several threads on this forum discussing use of
p-hydroxybenzaldehyde which after hydrogenation, provides rheosmin, "raspberry ketone." Without crunching the numbers, there looks to be an excess of
NaOH since the phenols are deprotonated initially and more beyond that is needed to catalyze the reactions.
Here is one such thread in prepub:
https://www.sciencemadness.org/whisper/viewthread.php?tid=12...
See also, http://sci-hub.tw/10.1007/s00897960034a
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@UC235, its interesting that in the Ozone lab prep they acidify the reaction medium after completion. I have found that this is only necessary with
only some of the phenolic aldehydes. When trying to prepare the bis-Aryllidene-acetones neutralisation of the alkali at the end of the reaction seems
to cause the vanillin compound to disappear!
Over the past few days I have tried to prepare the di-substituted acetones from anisaldehyde, vanillin, cinnamaldehyde, salicylaldahyde and
4(N,N-dimethylamino)benzaldehyde. Anisaldehyde and cinnamaldehyde appear to have worked well and gave copious yields of yellow crystalline material,
dimethylaminobenzaldehyde gave a poor yield that resembles the mono-substituted compound as red platy crystals, Salicylaldehyde gave very little until
a considerable excess of acetone was added and after several hours neutralised gave large deep yellow platy crystals presumably of the monosubstituted
compound and vanillin gave a little beautiful lemon yellow crystalline ppt but on partial neutralisation the ppt vanished and nothing could be
recovered.
Some test tude scale experiments I have carried out suggest that it may be better to dissolve the vanillin in a small excess of NaOH solution and then
add excess acetone to get the mono-substituted product.
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
The red 4-(N,N-dimethylamine)benzalacetone (photo above) is deep garnet when recrystallised but forms a colourless hydrochloride. This pH indicator
function was a bit of a surprise as was the depth of colour in neutral or weakly acid (acetic acid) So I was wondering if I may have got some other
type of condensation, particulalry as the same compound forms irrespective of the ratio of reactants?
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Has anyone ever tried this reaction with salicylaldehyde instead of benzaldehyde? There is a paper in the J. Biol. chem. which describes a test of
acetone using salicylaldehyde (J. Biol. Chem.; 1916; v27; p209 Colorometric Determination of Acetone in Urine; F A Csonka) where the salicylaldehyde
is in large excess and the a red colour develops; Csonka clear thought that colour was due to a bis-(2-hydroxybenzal)acetone from his proposed formula
but presents no actual data to support this.
I tried to prepare 2-hydroxybenzal acetone from excess acetone, salicylaldehyde and sodium hydroxide solution and obtained a straw yellow coloured
crystalline material. When I tried to recrystallise it from isopropanol (which appears to be the best solvent) pale green crystals formed leaving a
brown filtrate. Under the microscope the crystals can be seen to be a mixture of colourless platy crystals that turn brown on exposure to air and pale
green glassy blocky tablets that appear to be quite stable. When treated with dilute (1M) sodium hydroxide the filtrate and the crystals dissolve and
give an intense scarlet red solution. It is not clear yet which of the crystals is responsible for the colour.
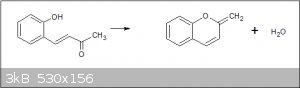
Do people think that this compound is a hydroxybenzal acetone? Is it possible that when isolated from the strong alkali that it undergoes
cyclotisation to give a chromane type compound?:-
|
|
|








