B.D.E
Hazard to Self

Posts: 97
Registered: 5-8-2019
Member Is Offline
Mood: Oscillating
|
|
ortho nitration of p-toluenesulfonic acid(QUESTIONS)
Hey there 
I just made some p-TsOH*H2O, and I'm hoping on nitrating its ortho position to make an easy precursor of o-toluidine and anthranilic acid.
Although the reaction seems like a "classic" textbook example for using SO3H as a protection group, I couldn't find anything about it
online(which made me a bit nervous).
My only experience with aromatic nitration was in the preparation of picric acid from salicylic acid(an easy "all the way" nitration of a
phenol).
So before trying anything, I hoped to hear some of the public wisdom in regard to the reaction conditions. My current plan is to dissolve the TsOH in
an excess of H2SO4(96%) and then gradually add ~1.1eq of NaNO3(s) while keeping the temperature on around 55C.
afterward if that won't work, I would try again at 70C and so on.
Does that sound right to you? also, is there a way to monitor the progress of the nitration?
---
The reaction scheme:
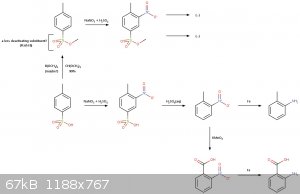
---
P.S I'm aware that some people are annoyed by others asking questions instead of simply trying and failing. But my time and reagents are limited, and
I just want to make sure I'm on the right track.
Also, I almost always report back on my attempts(usually with pictures), so I do "give back" in a way.
Thanks ahead, B.D.E
|
|
|
Tsjerk
International Hazard
Posts: 3022
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I can't find anyone reporting anything on this procedure either, but I guess it could work, or at least the nitration should work. But do keep the
possibility in mind the nitration runs via nucleophilic substitution, in which the para sulfonic group can be the leaving group as well as the ortho
hydrogen. So I wouldn't be surprised if there would be at least some p-nitrotoluene in the product.
A good first indicator of p-nitrotoluene in o-nitrotoluene would be the melting point.
Have you seen the HNO3 in DCM nitration of toluene? Chemplayer has a video on it on Bitchute. I'm planning on doing this reaction soon, I should get some technical toluene, KNO3 and DCM soon.
Chemplayer says he couldn't get the product to freeze at -12 (mp o-nitrotoluene ~ -10).
[Edited on 14-6-2020 by Tsjerk]
|
|
|
njl
National Hazard
Posts: 609
Registered: 26-11-2019
Location: under the sycamore tree
Member Is Offline
Mood: ambivalent
|
|
I just tried the dcm nitration and I honestly have never seen something so beautiful. No thermal runaway, no ice, reactants are easy to dry, barely
any fuming, easy workup. Just all around good experience.
|
|
|
B.D.E
Hazard to Self
Posts: 97
Registered: 5-8-2019
Member Is Offline
Mood: Oscillating
|
|
Quote: Originally posted by Tsjerk  | Have you seen the HNO3 in DCM nitration of toluene? Chemplayer has a video on it on Bitchute. I'm planning on doing this reaction soon, I should get some technical toluene, KNO3 and DCM soon.
Chemplayer says he couldn't get the product to freeze at -12 (mp o-nitrotoluene ~ -10).
[Edited on 14-6-2020 by Tsjerk] |
Wow, the DCM nitration sounds awesome. I think I'll give it a try instead.
| Quote: Originally posted by njl | | I just tried the dcm nitration and I honestly have never seen something so beautiful. No thermal runaway, no ice, reactants are easy to dry, barely
any fuming, easy workup. Just all around good experience. |
And that settles it - I'm definitely gonna try it 
May I ask for how long you kept the reaction going, at what temperatures(~) and what was your recovered yield?
---
Just as a side note, I'm actually quite glad I didn't had to use all of my beautiful white pTsOH:

I get that it might look ordinary to some of you, but for me this is the first time it didn't ended up as a brownish sludge. So I quit happy I get to
keep it for a while 
[Edited on 14-6-2020 by B.D.E]
|
|
|
njl
National Hazard
Posts: 609
Registered: 26-11-2019
Location: under the sycamore tree
Member Is Offline
Mood: ambivalent
|
|
I don't remember the yield but I left the solution of dcm/HNO3/toluene stirring overnight (probably 12 hours total).
|
|
|
chemplayer...
Hazard to Others
Posts: 191
Registered: 25-4-2016
Location: Away from the secret island
Member Is Offline
Mood: No Mood
|
|
Wish we had had the set-up to characterise the product of this reaction a bit better. Definitely an ortho-rich product though at the very least. Would
like to see just how selective the reaction is.
Please be a little bit careful as too high a concentration of nitric acid in DCM has been reported to be unstable!
|
|
|
B.D.E
Hazard to Self
Posts: 97
Registered: 5-8-2019
Member Is Offline
Mood: Oscillating
|
|
| Quote: Originally posted by njl | | I don't remember the yield but I left the solution of dcm/HNO3/toluene stirring overnight (probably 12 hours total). |
could you estimate it roughly?(i.e below 50%? above 50%? about 50%?)
because chemplayer ran the reaction for about 2 hours, and got a yield of 29%. So if the yield is still below 50% after an additional 10 hours, I'll
probably make do with just 3-4 hours instead(the reactants are cheap enough).
| Quote: Originally posted by chemplayer... | Wish we had had the set-up to characterise the product of this reaction a bit better. Definitely an ortho-rich product though at the very least. Would
like to see just how selective the reaction is.
Please be a little bit careful as too high a concentration of nitric acid in DCM has been reported to be unstable! |
Thanks man! I understand you frustration about not being able to better analyze the product. And thanks for the warning(I also saw the warning in your
video).
|
|
|
njl
National Hazard
Posts: 609
Registered: 26-11-2019
Location: under the sycamore tree
Member Is Offline
Mood: ambivalent
|
|
I think it was in the neighborhood of 60+ percent, but this was before I got a good scale and I can't speak to the purity as I just did the usual
freezing.
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@B.D.E; I am currently trying to prepare 2-amino-4-chlorophenol-6-sulphonic acid which is an azo dye intermediate. The theory is easy, the practical
details are a little more difficult. I have found that at room temperature nitration of 4-chlorophenol-2-sulphonic acid is rapid even in 85% sulphuric
acid with sodium nitrate solution but there is significant displacement of the sulphonic acid group by a nitro group to give a mixture of the desired
product plus 2,6-dinitro-4-chlorophenol and 2-nitro-4-chlorophenol. The mixture is easily separated but represents significant loss. However, if
nitration is carried out at about 0 C the desired 2-nitro-4-chlorophenol-6-sulphonic acid is obtained in good yield with minimal nitrophenols.
I suspect that you will find that low temperature nitration will favour o-nitration over displacement of the sulphonic acid group in the p-position.
Incidently I came a cross an article (that I am currently trying to locate) that suggests that nitrous acid tend to favour displacement of sulphonic
acid groups in aromatic compounds so purity of your nitric acid may be an issue. I have also found that the use of sodium nitrate is best avoided
because the separation of sodium salts at low temperatures makes the mixture very difficult to stir and therefore make temperature control difficult.
I don't know how easily toluene sulphonic acid will nitrate, I suspect less easily than my chlorophenol sulphonic acid so you may need conc sulphuric
and conc nitric acid but try and keep the temperature low.
Let us know how you get on.
|
|
|
B.D.E
Hazard to Self
Posts: 97
Registered: 5-8-2019
Member Is Offline
Mood: Oscillating
|
|
| Quote: Originally posted by Boffis | @B.D.E; I am currently trying to prepare 2-amino-4-chlorophenol-6-sulphonic acid which is an azo dye intermediate. The theory is easy, the practical
details are a little more difficult. I have found that at room temperature nitration of 4-chlorophenol-2-sulphonic acid is rapid even in 85% sulphuric
acid with sodium nitrate solution but there is significant displacement of the sulphonic acid group by a nitro group to give a mixture of the desired
product plus 2,6-dinitro-4-chlorophenol and 2-nitro-4-chlorophenol. The mixture is easily separated but represents significant loss. However, if
nitration is carried out at about 0 C the desired 2-nitro-4-chlorophenol-6-sulphonic acid is obtained in good yield with minimal nitrophenols.
I suspect that you will find that low temperature nitration will favour o-nitration over displacement of the sulphonic acid group in the p-position.
Incidently I came a cross an article (that I am currently trying to locate) that suggests that nitrous acid tend to favour displacement of sulphonic
acid groups in aromatic compounds so purity of your nitric acid may be an issue. I have also found that the use of sodium nitrate is best avoided
because the separation of sodium salts at low temperatures makes the mixture very difficult to stir and therefore make temperature control difficult.
I don't know how easily toluene sulphonic acid will nitrate, I suspect less easily than my chlorophenol sulphonic acid so you may need conc sulphuric
and conc nitric acid but try and keep the temperature low.
Let us know how you get on. |
That's very useful man. Much thanks!
I already tried the dcm method and it seemed to went well. unfortunately I accidentally spilled the reaction flask just before evaporating the dcm. So
I redoing it now :S
I actually also planned on trying the dcm method with pTsOH instead of toluene. But after reading your comment I'm now reconsidering my options.
Also, there's still the problem of assessing the purity and regioselectivity of any of the method's products. So if anyone can think of way to even
compare these aspects between the different batches - please share :)
[Edited on 16-6-2020 by B.D.E]
|
|
|
Tsjerk
International Hazard
Posts: 3022
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
Melting point of compounds is a nice way to determine whether it is pure. Mixtures however often are hard to determine by meltingpoint.
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I would be interested to hear how you got on with the DCM solvated nitration. I have never tried it but I have read a couple of papers on it.
If you successfully nitrate p-toluene sulphonic acid the nitro-sulphonic acid will probably be water soluble and it may take a bit of investigation to
find a suitable means of isolating it. However if you use mixed acids and don't use excess nitric acid you may be able to dilute the reaction mixture
and then reflux in the excess sulphuric acid to cleave the sulphonic acid group.
|
|
|
B.D.E
Hazard to Self
Posts: 97
Registered: 5-8-2019
Member Is Offline
Mood: Oscillating
|
|
Well, the DCM nitration didn't went very well to say the least.
I started by mixing 0.2n of KNO3(oven dried and crushed in a mortar) with 0.188n of H2SO4(96.1%). After some stirring
a thick "gel" had formed.
I added ~75gr of DCM and the gel transformed into a "breakable" solid. I broke the solid into small pieces and allowed the mixture to stir in an ice
bath.
After about an hour, I weighted 0.22n of toluene and diluted it with additional ~25gr of DCM. I added the toluene/DCM mixture into the
HNO3/DCM solution slowly(3ml at a time), and the mixture had turned orange-red. After the addition was over, I left the mixture to stir for
about 7-8 hours in the ice bath(ice bath: T0~0C, Tf~18C).
After 7-8 hours I poured the content of the flask into a a sinter funnel and filtered the solids off. I transferred the filtrate into a separatory
funnel, washed with ~20mL of dH2O, and removed the top aqueous layer. I then added about 20mL of saturated K2CO3
slowly(to avoid boiling off the DCM), afterwards the DCM layer was on top. I decanted both layers and this is what I was left with:

I connected a distillation apparatus to the "DCM layer" and distilled everything that came under 105C. Afterwards I was left with a very small amount
of crude product.
I transfered the product via pipette into a test tube, washed it few times with water and dried it with some silica gel. The dry yield was only 3.5gr.
Due to the low yield, I decided to do the same workup(i.e. distillation>washing>drying) on the aqueous layer, but only got additional 0.5gr.
Total overall yield was 4gr(=15.5% from HNO3).

The product remains liquid at -13C.
Plausible reasons for the low yield:
the reaction temperature was too low.
the reaction needed more time to complete.
the excess toluene I've used was a bad idea for some reason.
Tomorrow I hope to try the mixed acid method(pTsOH + H2SO4 + NaNO3 at 0C).
Good night.
[Edited on 18-6-2020 by B.D.E]
|
|
|
Tsjerk
International Hazard
Posts: 3022
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
With a bit of luck I can try this reaction tomorrow, I will let you know what happens.
I'm not sure yet whether I will separate the DCM from the sulfuric acid/KNO3 before adding the toluene. The paper Chemplayer refers to in the video
appears not to do so. Maybe I will extract the HNO3 three time with DCM and pool the extracts before attempting the nitration.
Are you sure you distilled of everything under 105 degrees? Toluene has a boiling point of 110...
|
|
|
B.D.E
Hazard to Self
Posts: 97
Registered: 5-8-2019
Member Is Offline
Mood: Oscillating
|
|
| Quote: Originally posted by Tsjerk | With a bit of luck I can try this reaction tomorrow, I will let you know what happens.
I'm not sure yet whether I will separate the DCM from the sulfuric acid/KNO3 before adding the toluene. The paper Chemplayer refers to in the video
appears not to do so. Maybe I will extract the HNO3 three time with DCM and pool the extracts before attempting the nitration.
Are you sure you distilled of everything under 105 degrees? Toluene has a boiling point of 110... |
Cool. I'm looking forward to see how it would play out for you.
As for the distillation - I basically placed everything inside an oil bath(T>140C) and distilled everything until the vapor temperature dropped.
I'm pretty sure the temperature never actually reached 110C.
[Edited on 18-6-2020 by B.D.E]
Also, I personally think that doing the reaction in "one-pot" is the best way to go. The KHSO4 breaks quite easily after adding the DCM,
and it shouldn't affect the reaction in any way.
[Edited on 18-6-2020 by B.D.E]
|
|
|
Tsjerk
International Hazard
Posts: 3022
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I had such a great idea... only the execution was a bit poor.
I started the reaction, everything looks fine. I will run it overnight and will report on the yield tomorrow.
My great idea: I extracted the HNO3 three times with DCM from the sulfuric acid/KNO3. I know exactly how much sulfuric acid and KNO3 I used, so I can
calculate how much acid would be left in the salty sulfuric acid/KHSO4/KNO3 mix when the extraction would be 100% efficient. I calculated how much
NaOH would be needed to neutralize that acid. I could then measure the pH of the "neutralized" mixture which would correspond with the HNO3 not
extracted with the DCM.
So I dissolved the salt mix in 500 ml of water and added the amount of NaOH... Only to realize there was a bit of DCM on the bottom of the beaker
which of course started to boil trowing 4/5th of the contents of the beaker all over the place... So no pH measured.
But tomorrow I will quenche the reaction with a known solution of bicarbonate to see how much nitric acid is left which together with the nitrotoluene
should sort of correspond with the amount of HNO3 extracted.
|
|
|
B.D.E
Hazard to Self
Posts: 97
Registered: 5-8-2019
Member Is Offline
Mood: Oscillating
|
|
| Quote: Originally posted by Tsjerk | I had such a great idea... only the execution was a bit poor.
I started the reaction, everything looks fine. I will run it overnight and will report on the yield tomorrow.
My great idea: I extracted the HNO3 three times with DCM from the sulfuric acid/KNO3. I know exactly how much sulfuric acid and KNO3 I used, so I can
calculate how much acid would be left in the salty sulfuric acid/KHSO4/KNO3 mix when the extraction would be 100% efficient. I calculated how much
NaOH would be needed to neutralize that acid. I could then measure the pH of the "neutralized" mixture which would correspond with the HNO3 not
extracted with the DCM.
So I dissolved the salt mix in 500 ml of water and added the amount of NaOH... Only to realize there was a bit of DCM on the bottom of the beaker
which of course started to boil trowing 4/5th of the contents of the beaker all over the place... So no pH measured.
But tomorrow I will quenche the reaction with a known solution of bicarbonate to see how much nitric acid is left which together with the nitrotoluene
should sort of correspond with the amount of HNO3 extracted. |
That's a cool idea, but I still think you can get away with just running the reaction in one-pot. In any case, I'm looking forward to hear
how it went down for you(hopefully you'll get better result than I did).
---
I tried the mixed acid method at T<5°C and it seemed to went pretty well. I used fuming white(ish) HNO3 with 96%
H2SO4. I also used a small excess of p-TsOH to avoid dinitro products(the excess will be later converted to
easy-to-remove toluene in the following desolfunation step).
The p-TsOH was dissolved in H2SO4, and a mixture of {HNO3/H2SO4} was added dropwise. the
nitration was rapid, as evidenced from the temperature rising with every addition of the{HNO3/H2SO4} mixture.
After the addition was complete, I sniffed the rxn vessel and couldn't detect the aromatic odor associated with nitrotoluenes(which was a good sign,
as it suggested that the product was mostly 2-nitrotoluene-4-sulfonic acid and not p-nitrotoluene).
I then added a few millilitres of water to the rxn flask, the temperature quickly rose to ~10°C and the reaction mix was partially solidified to a
slush-like consistency. I'm not sure if that was the hydrate of the leftover p-TsOH or possibly the hydrate of our predicted product. Anyway,
I struggled to vacuum filter it all out and was left with 2 solutions:
a) filtrate/H2O
b) solids/H2O
(looking back I definitely added way too much water to both of the solutions)
I refluxed both solutions for an hour, and a strong aromatic odor had appeared(it was consisted with the smell of my DCM-nitration product).
I then added a few drops of phenolphtaleine and slowly neutralized both solutions with concentrated K2CO3(aq)
solution. lots of foaming had occurred and a white solid had appeared. At some point of the neutralization, the solutions had turned from mostly-clear
to a distinct yellow, and shortly after to reddish-pink(due to the phenolphtaleine added).
I filtered off the newly-formed-solids from both solutions, and was left with two homogeneous solutions. This is unfortunate as at this point
the o-nitrotoluene should have been phase separated from the aqueous layer(ref).
I steam distilled some of the water to see if it would make a difference, but it was in vain.
---
A quick discussion:
It was clear that some sort of nitration took place. Given that the reaction flask had no smell at the end of the nitration, it is unlikely that
product was a none-sulfonated nitrotoluene(either o or p isomers). Thus, it was most likely the
2-nitrotoluene-4-sulfonic acid we expected to make.
After doing a quick search online, I came to believe that the problem lies in the desulfonation stage. Apparently, the desolfunation reaction requires
high concentrations of at least 65%(wt) H2SO4 in order to be somewhat time efficient(ref). I instead used an over-diluted aqueous solution for a relatively short period of time(under the false assumption that any acidic-aqueous
environment would do).
---
I'm planning on trying the reaction again very soon. This is also why I didn't bothered adding pictures or mentioning the exact amounts I've used(I
would do so after the reaction will succeed).
[Edited on 20-6-2020 by B.D.E]
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@B.D.E.; very interesting results. The solid that separated when you diluted the reaction mxture; did you keep itand investigate it? Is it organic?
You talk about two solutions, one is the filtrate but the other? Did you dissolve the solid in water, I didn't quite understand here?
Did you investigate the "newly formed" solid? Is it organic and could it be 2,6-dinitrotoluene (lower volatility and slight odour) as it is presumably
insoluble in water/dilute acid. By the way, o-nitrotoluene and its sulphonic acid are capable of undergoing internal redox reactions to give
anthranilic acid and anthranil derivatives (see Richter, volII).
From my experience aromatic nitro sulphonic acids are very soluble and so are most of their alkali and alkaline earth salts. Maybe try neutralising a
part of the reaction mixture with calcium hydroxide or carbonate, stand for a few days to allow the gypsum to crystallise (this can be very slow) and
filter of the soluble salts and evaporate down to crystals. I have observed that the solubility gradients of Ca sulphonates are often small so its
often difficult to recover them by cooling the saturated solution; its usually a steam-bath job. The free acid can then probably be derived from this
with dilute sulphuric acid and filtering off the gypsum or you may be able to de-sulphonate it directly making an allowance for the extra sulphuric
acid needed to liberate the free acid.
You can also use sodium carbonate, most of the resulting sodium sulphate actually crystallises out as impressive large colourless crystals that are
easily removed and washed. You are right about the strength of sulphuric acid to desulphonate a sulphonic acid, these reaction tend to be equilibrium
reaction. In the case of m-xylene for instance 65-70% acid will sulphonate the m-xylene so you have to continuously remove the xylene liberated by
steam distillation as it forms.
Have you looked for data on 2-nitrotoluene sulphonic acid and salts?
|
|
|
B.D.E
Hazard to Self
Posts: 97
Registered: 5-8-2019
Member Is Offline
Mood: Oscillating
|
|
As for the solid that formed after adding water to the nitrated solution, here how the mixture looked like after the precipitation occurred:
 
(I know it doesn't really help with anything, but it definitely can't hurt ).
I struggled filtering all of the liquids out of the solid, so I settled on just filtering as much as I could(probably most of it), before adding some
arbitrary amount of dH2O directly into the sinter funnel to dissolve whatever had left. The solid dissolved quite easily to create said
solution "(b)"(from my previous post). I also kept the filtrate from before adding the water(aka solution "(a)"), and ran all of the following workup
steps on both solutions separately.
As for the solid that formed while neutralizing said solutions, I didn't kept it nor investigated it at all, as I was sure it was just
K2SO4(at the time). I did however washed it out of the sinter funnel and it seemed to dissolved well in water.
As for the reduction to anthranilic acid - I'll be sure check your reference soon and maybe try adding CuSO4 to samples of the solutions I
was left with, in order to see if I can ppt any copper anthranilate. Much thanks for this info.
#Update - I added some Cu(NO3)2 and no green ppt had formed.
The calcium method sound a bit too messy for me. Do you really think it's necessary to isolate the sulfonated intermediate to such an extent before
the desulfonation? If so, I hope that it's indeed possible to isolate it as an hydrate directly after the nitration(it's seems as if its ortho isomer does form an hydrate, so there's still hope in my heart).
As for "data about 2-nitrotoluene sulphonic acid and salts", I did looked it up on chemspider and pubchem, but couldn't find anything that
seemed relevant. I also checked Ullmanns and Vogel, but couldn't find anything useful on there as well. (I only searched for the
free acid).
Much thanks for the reply and the willing to help, I really do appreciate it
[Edited on 20-6-2020 by B.D.E]
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@B.D.E.; I checked out some old chemical dictionaries and got some references to the nitration of toluene-4-sulphonic acid. I can't track down the
first reference but it must be before 1900 since by 1901 2-nitrotoluene-4-sulphonyl chloride was being used in experiments into the esterification of
phenol without a description of its preparation so was clearly considered a staple reagent. The most useful paper I found was:
Fichter & Bernoulli; 1909; The electrolytic reduction of 2-nitrotoluene-4-sulphonyl chloride; Berichte; v42; p4309.
In this paper they describe the preparation of the starting material from sodium toluene-4-sulphonate via direct nitration of the salt. I have already
found that it is usually better to nitrate sulphonate salts rather than try and isolate the free sulphonic acid.
|
|
|
Tsjerk
International Hazard
Posts: 3022
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I first cleaned my technical toluene by stirring 100 ml with 15 ml concentrated H2SO4 at RT for about an hour. When the sulfuric acid wasn't
becoming more yellow I separated the acid and the toluene in a separatory funnel and washed the toluene with a sodium carbonate solution.
I dried the toluene with magnesium sulfate and filtered the toluene and stored it overnight over a bit of magnesium sulfate.
I slowly added 0.4 mol (40 gr) of oven dried KNO3 to 0.466 mol of cooled H2SO4 on an ice bath. Once everything was added, keeping the
temperature below 30 degrees, a thick paste formed. The paste couldn't be stirred with a magnetic stirrer anymore and was stirred by hand.
The HNO3 was extracted with 75 ml DCM. At first the sticky mass is difficult to stirr and extraction seems slow, as no nitric acid fumes are produced
at first.
After some manual stirring (stainless steel spoon is fine in this mixture) the mass becomes more loose, and vapors start coming off.
After about ten minutes the DCM was pored off and the now much more crystalline mass is extracted with 50 ml DCM. Magnetic stirring is still not
possible,
but manual stirring becomes easier. The mass becomes even more crystalline. The same procedure is repeated with 50 ml more DCM.
After manually squeezing out as much DCM as possible I dissolved the left over solids in 500 ml of water. If the extraction is around 100% efficient
there should
be 0.066 mol of H2SO4 left and 0.4 mol of KHSO4. I took 100 ml of the solution and added 3.7 grams of NaOH (0.4 + 0.066 mol / 5). I should have added
4.26 ((0.4 + 0.066 + 0.066) / 5)
which should have given a neutral solution when the extraction was 100% efficient... But OK, 3.7 grams should have given an pH of ~1.8. My pH paper is
not that accurate
at low pH apparently as the pH was 1, and still 1 after diluting 10 times... after another 10x dilution the pH was around 4. Which calculates back to
a pH around 2
after the neutralization with NaOH. So probably most HNO3 was extracted with the DCM.
After nitration I needed 10 grams of NaHCO3 to neutralize, which corresponds with 0.12 mol HNO3. So 70% of the HNO3 should be in either the
crystalline cake or bound
as nitrotoluene.
I added 0.4 mol of the cleaned and dried toluene, both toluene and DCM/HNO3 were cooled on ice, but there was no exothermic reaction observed. The
addition of toluene
produced a dark red solution, which became orange when warmed to RT. After getting to RT there was a bit of exothermic reaction, just enough to make
the DCM reflux a little.
After 24 hours at RT, the reaction was quenched with sodium bicarbonate. Bubbling was observed, and the after bubbling stopped the reaction was washed
with water. The organic layer
was washed with water and dried with magnesium sulfate. DCM was distilled of and 100 ml was recovered. Toluene was removed under vacuum, at which the
toluene boiled at 50 degrees.
Yield: 11.5 gr, 21% based on the nitrate. I did my best to evaporate all toluene, but I couldn't get the product to freeze at -16. My guess is that
there is still a bit of toluene left.
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@Tsjerk; In the paper that describes this method I am sure they reckoned that extraction was about 80% so your extraction seems reasonable but the
yield on nitrotoluene seems low. I try and dig out the original paper.
@B.D.E.; I have delved deep into the online archives and found some really interesting stuff. There is a great deal of information on toluene
chemistry in Thorpe's Dictionary of Applied Chemistry vol VII (1927 edition) and very specifically there is a statement that toluene sulphonic acid
can be hydrolysed by steam at 160 C. There is no mention in any reference I can find of the hydrolysis of 2-nitrotoluene-sulphonic acid or for that
matter of the 2,6-dinitro sulphonic acid. These acids seem to undergo intramolecular reactions between the nitro and methyl groups rather readily. Is
your low freezing point surprising? I am sure some p- and m- isomers are still present but if no p- isomer crystallised out then maybe you have an o-
isomer rich mixture.
I also found the original reference to the nitration of toluene-4-sulphonic acid with mixed acids: Engelhardt & Bek; Zeitschrift fur Chemie; 1869;
series 2; vol 5; p209. Both this reference and Thorpe (full series of 1927 edition 7 volumes) can be down loaded from Archive.org.
I also discovered that you can 2-nitrate the -4-sulphonamide which is readily available in the UK
|
|
|
B.D.E
Hazard to Self
Posts: 97
Registered: 5-8-2019
Member Is Offline
Mood: Oscillating
|
|
@Tsjerk Great write-up man. I must admit it's a bit disappointing to hear that even though you seemed to have done everything right - you still got a
rather low yield.
To be fair, the reference we all followed(ref[right?]) never really nitrated toluene. So at least as far as I know, the reaction might just not work well with toluene under those
condition.
---
@Boffis Thanks for all the info
Unfortunately some of the references you gave are in German. I also find it quite hard to read old papers as such(freaking 1869 man!  ). It was cool though to see how much work people have put into some of these
papers(especially the detailed hand drawings). ). It was cool though to see how much work people have put into some of these
papers(especially the detailed hand drawings).
As for the self reduction/oxidation, I have found this:

ref: 978-0-470-03403-3(ISBN) page 113
Meaning that the oxidation probably requires a catalyst in order to be rapid. I didn't dag too much into it, but from what I could found it didn't
seems likely that this should be an issue in my case.
As for your comment about Aryl-sulfonate salts being easier to isolate than their free acids - I would have agreed with you if pTsOH wouldn't
have been so easy to isolate as an hydrate directly from its mother solution(i.e the solution in which it was prepared). After all, there isn't a real
advantage in nitrating pTsONa(for instance) over simply nitrating pTsOH(obviously the salt would be converted to the free acid the moment you'll add
the mixed acid).
As for this comment:
| Quote: | | Is your low freezing point surprising? I am sure some p- and m- isomers are still present but if no p- isomer crystallised out then maybe you have an
o- isomer rich mixture. |
-I still haven't desolfunated the intermediate, so at least for the time being I'm yet to have checked a freezing point.
The only thing I did since my last update was to completely evaporate all water from the solutions I previously made("a" and "b"). This is what I got:

(b) - on the right ; (a) - on the left
With a bit of luck, one of those should contain 2-nitrotoluene-4-sulfonate(potassium salt).
I planing to get back into it next Sunday(28).
Hopefully I would be able desulfonate the intermediate and get the promised o-nitrotoluene.
[Edited on 23-6-2020 by B.D.E]
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
OK here's a translation of the most useful paper from Zeit. f. Chem.. I have only translated the bit by Bek but its pretty useful. There are a few
things I wasn't sure about like what the hell is metabenzylsulphohydrate (Metabenzylsulfhydrats in the original)? There are also a few clear errors,
they don't seem to have appreciated that barium was divalent and Na/K monovalent so the formulas are a bit suspect and the hydration states of the
barium and lead salts are clearly wrong. For instance the writer claims that the theoretical water content of the monohydrate is 5.95% but I
calculated about 3% so it looks like the salt may be a dihydrate.
Attachment: Nitrotoluene-4-sulphonic acid and derivatives Zeit.f.Chem Engelhardt & Bek 1869 EngTrans.docx (20kB)
This file has been downloaded 295 times
There is a lot of amazing chemistry in that old German literature, particularly before about 1930 since most of the chemicals and equipment they used
ares now readily accessible (often from ebay with the exception of Oleum  ). ).
The clip you posted above re-oxidation of nitrotoluene sulphonic acids refers to intermolecular reactions with external oxidation. Under certain
conditions 2-nitrotoluene and its 4-sulphonic acid will undergo intramolecular oxidation where the nitro group will oxidise the methyl group. The
yields are often not good enough to make these preparatory methods because there are often a lot of side reaction but you need to know about them if
you are going to apply fairly brutle reaction conditions to something like 2-nitrotoluene. I'll see if I can find some of the references (probably in
German too!) These reactions do appear to occur mostly in alkaline solutions though.
|
|
|
B.D.E
Hazard to Self
Posts: 97
Registered: 5-8-2019
Member Is Offline
Mood: Oscillating
|
|
| Quote: Originally posted by Boffis | OK here's a translation of the most useful paper from Zeit. f. Chem.. I have only translated the bit by Bek but its pretty useful. There are a few
things I wasn't sure about like what the hell is metabenzylsulphohydrate (Metabenzylsulfhydrats in the original)? There are also a few clear errors,
they don't seem to have appreciated that barium was divalent and Na/K monovalent so the formulas are a bit suspect and the hydration states of the
barium and lead salts are clearly wrong. For instance the writer claims that the theoretical water content of the monohydrate is 5.95% but I
calculated about 3% so it looks like the salt may be a dihydrate.
There is a lot of amazing chemistry in that old German literature, particularly before about 1930 since most of the chemicals and equipment they used
ares now readily accessible (often from ebay with the exception of Oleum ).
The clip you posted above re-oxidation of nitrotoluene sulphonic acids refers to intermolecular reactions with external oxidation. Under certain
conditions 2-nitrotoluene and its 4-sulphonic acid will undergo intramolecular oxidation where the nitro group will oxidise the methyl group. The
yields are often not good enough to make these preparatory methods because there are often a lot of side reaction but you need to know about them if
you are going to apply fairly brutle reaction conditions to something like 2-nitrotoluene. I'll see if I can find some of the references (probably in
German too!) These reactions do appear to occur mostly in alkaline solutions though. |
Wow man, thank you very much for translating this for me. I see now what you're mean.
Oh well, you win some and you lose some I guess "^_^
Nonetheless, just because I already got this far, I felt as it will be a waste not to at least try the reaction myself. So I made a 80%
H2SO4(aq) solution and heated it to ~170C for around 1.5 hours with s.stirring. After letting the mixture cool, I poured it into
an excess of water and carefully sniffed the dilute homogeneous solution - I couldn't identify any trace of the aromatic aroma associated with
nitrotoluene(nothing at all).
Due to me being at the midst of another project, I decided not to research the product/products any further by isolation and thus poured the solution
down the drain.
Thanks again boffis, you are a very nice guy and the info you sheared helped a lot
|
|
|









