Fery
National Hazard

Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
allyl phenoxyacetate - pineapple scent
I prepared allyl phenoxyacetate by esterification of allyl alcohol and phenoxyacetic acid. This is so far the best pineapple scent I have ever made, my favorite, No. 1.
No. 2. ethyl hexanoate
No. 3. allyl penatanoate
Cou if you are reading this I definitely suggest you to prepare this ester into your incredible ester collection together with ethyl 2-ethylhexanoate which differs a lot from usual floral/fruit scents of most common esters.
To 250 ml RBF I placed phenoxyacetic acid 0,25 mol 38,0 g (Molar mass = 152.15 g/mol). My product was cotton like of big volume / needles so it almost
filled the whole flask.

Then allyl alcohol added 0,5 mol 29,0 g (Molar mass = 58.08 g/mol), but I used fraction boiling 88-96,5 C (approx 85% allyl alcohol concentration) so
the molar mass >0,4 mol so still >60% excess. Because using Dean-Stark trap apparatus the small amount of water does not mind and comparable
amount of water will be generated in the reaction (I just wanterd to keep the anhydrous allyl alcohol for reactions where water causes trouble, like
transesterification). Some of allyl alcohol will be lost in water trapped in the apparatus as it is not pure water but with cca 18% of allyl alcohol.
If 10 ml of "water" trapped it will contain cca 2,0 g of allyl alcohol so the allyl alcohol used for esterification is still in excess.
Added 0,25 ml 96% H2SO4 and 5 small glass balls as boiling stones.
Attached Dean-Stark trap apparatus, condenser, gas adapter to which a hose leading outside of building was attached (allyl alcohol is powerful
lachrymator, few mg leak and you are crying like when crushing garlic / cutting onion). Also all ground glass joints greased well to avoid any leak.
The RBF was slightly heated on oil bath so the reactants dissolved, mixed well and the mixture started to slowly reflux at which point I turned the
flame off and I let it to reflux so that the vapor condensed on the flask walls and on the arm of the Dean Stark trap apparatus so all of the
condensate returned back into the flask and did not fill the trap. Then I let it to cool down for 1 hour. This was just to create some ester and
dissolve reactants as they could be poorly soluble in hexane.

When the mixture was cool (certainly at least 10 C less than the low range of boiling point of the hexanes/heptanes mixture which form azeotropes with
even less boiling points) I added 30 ml of distilled medicinal petrolether (fraction boiling 60-80 C, medicinal petrolether used in my country is
always hydrogenated fraction so without unsaturated hydrocarbons) from the top of condenser and started to heat the reaction again.
After 30 minutes 5 ml H2O collected (0,25 mol) but it certainly contained cca 18% of allyl alcohol and also allyl alcohol used was not water free
(water content cca 15% = cca 4,5 ml of H2O). After 60 minutes 10 ml H2O collected. After 90 minutes still 10 ml which should be cca 8 g H2O (cca
0,45-0,5 mol) and 2 g of allyl alcohol. Heating turned off, water drained off and also all hexane distilled out (oil bath still hot) and drained from
the trap. I should let the hexane in the mixture (would make workup easier). When you repeat this experiment, drain only water after cooling down when
the reflux stops and let as much of hexane in the reaction flask as possible.

Then it was let to cool down to room temperature, transferred into separatory funnel, added 100 ml of water - the ester stayed as BOTTOM layer
(density 1,1 g/ml), after shaking with water it emulsified and did not want to separate so 20g NaCl added (you can directly wash it with 20% NaCl
solution) then it separated well as UPPER layer. Or completely avoid this when having hexane (low density solvent) still with it.
Then washed 2x with 50 ml of 8% NaHCO3 here the ester stayed as BOTTOM layer (density of 8% NaHCO3 seems to be lower than the density of the ester) -
after shaking the water phase was diluted to half by adding 50 ml of water to support better separation.
Then washed 2x with 50 ml 20% NaCl (product in UPPER layer), at the end the emulsion did not separate so I had to add 20 ml of the hexane which I
previously drained from Dean-Stark trap apparatus and only then the emulsion broke and separated sharply. Definitely do not remove the hexane from the
trap as I did. Let it there. I wanted to make workup faster and easier (avoid removing hexane from the product before vacuum distillation) but this
idea revealed to be wrong way to go.

Dried with anhydrous Na2SO4 and put into freezer overnight - during freezing temperatures -18 C a lot of hexane separated as an upper layer
(colorless, bottom dirty ester layer yellow-orange), on warming up the layers mixed.
It was still not clear so it was decanted into new flask and added fresh anh. Na2SO4 and put on magnetic stirrer, after 2 h of rapid stirring it was
clearer and after 4 hours of rapid stirring it was finally crystal clear. Without rapid magnetic stirrer it was impossible to properly dry out the
product. Maybe no problem as hexane should remove water when distilling it out of the ester.
  
Then all hexane was distilled out without messing condenser going to be used for final vacuum distillation, just few adapters and a flask in cold
water bath were enough. This was done in oil bath, the distillation started when T of oil reached 120 C and was completed when T of oil raised to 208
C, at which the distillation of hexanes/heptanes/azeotropes ended. Boiling point of the product is above 250 C at atmospheric pressure.
  
Then it was vacuum distilled from the same flask to which the rest of vacuum distillation apparatus was attached.
 
My manometer is not capable to measure so fine pressure changes so I was unable to compare the pressure difference with actual atmospheric pressure
(actually certainly low atmospheric pressure conditions according the local weather). The distillation started when the T of oil reached 174 C and the
T in distillation head 121,5 C. Few ml (cca 2-3 ml) was collected as a forerun which was drained down from Anshutz-Thiele separator and when the T in
head was 122,0 C the main fraction was collected, most of it distilled at 123,0 C.
  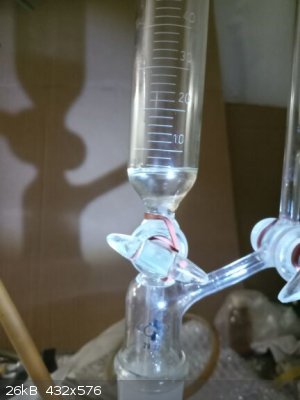
  
 
When the T in head rose to 124,0 C I was prepared to finish the distillation and when I managed to stop it the T in head already reached 125,0 C
(during the whole distillation the T was stable for long time at 123,0 C and at the end the increasing was very fast in few seconds) at which I
allowed to slowly enter air into the apparatus which effectively stopped the distillation immediately.
 
And here the product, final yield yield 38,2 g (0,20 mol, 80%) cca 34,5-35 ml, I weighed it on scale after bottling it to have more precise result
than the volume of liquid (the adapter graduation is only approximate). Also cca 2-3 ml of forerun in the test tube and cca 2 ml of dark residuum in
distillation flask covered with white stir bar.
  
and some info from internet:
http://www.thegoodscentscompany.com/data/rw1028371.html
Molecular Weight: 192.2
Specific Gravity: 1.10000 to 1.11000 @ 25.00 °C.
Refractive Index: 1.51200 to 1.51900 @ 20.00 °C.
Boiling Point: 100.00 to 102.00 °C. @ 1.00 mm Hg
Boiling Point: 265.00 to 266.00 °C. @ 760.00 mm Hg
|
|
|
njl
National Hazard
Posts: 609
Registered: 26-11-2019
Location: under the sycamore tree
Member Is Offline
Mood: ambivalent
|
|
THIS is the kind of content I love from the forum. Good shit my man. Obscure ester synthesis is something I can get behind 
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Thx njl, the first attempt worked flawlessly (synthesize the phenoxyacetic acid first, then perform its Fisher esterification).
My second idea was:
monochloroacetic acid + allyl alcohol -> allyl monochloroacetate (Fisher esterification)
allyl monochloroacetate + K phenolate -> allyl phenoxyacetate
And the third idea:
Na phenoxyacetate + allyl bromide.
Also the yield was surprisingly quite good for such small scale, at bigger scale it could be even higher. Some surprises happened during the work
again as always (emulsification of the ester in separatory funnel, unwillingness to dry the ester resting over drying agent in a peace) so I found
some improvements how to overcome them. The scent is really nice, pure natural, no traces of anything chemical (solvent like) - very likely due to
quite high boiling point.
In this synthesis I also used only very little of catalyst (0,25 ml H2SO4 which is cca 5 drops for 0,25 mol of ester) as recently in n-heptyl butanoate synthesis I observed a carbonisation and the catalyst stayed undissolved at the bottom of the reaction flask when used 5x more
catalyst (5 ml H2SO4 for 1 mol ester scale)... the smaller amount of catalyst was now sufficient as after 1 hour the esterification was completed (10
ml in Dean-Stark trap apparatus after 60 minutes and the same after 90 minutes).
|
|
|
Pumukli
National Hazard
Posts: 686
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
I love these times, when quality work is being done every day and gets published in the organic chemistry section of the forum.
Congratulations Fery!
I have some phenoxy-ethanol and was wondering if you were too busy to prepare a few interesting (reverse) esters from it.
I have the well-known problem (so many compounds but so little time) so guess what: I'd be willing to send you a sample of this compound, entirely
free of charge if you lived somewhere in the EU. (I think you do.) I only want to see a similar synthesis description materialize from this.
By reverse I mean the following: you prepare say phenoxy-acetic-acid ethyl ester AND also prepare the acetic acid ester of phenoxy-ethanol. Then
compare the smell of the two and tell us if they were different or not and if they were: what was the difference!
What do you think?
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Well done, another nice (smelling ) synthesis. Like Pumukli says, its good to
see so much practical chemistry, particularly organic chemistry, being done these days. A few years ago practical reports had almost disappeared.
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi Pumukli, thank you for your offer. You have it similarly to me... allyl alcohol prepared already 2 months ago and the acid 3 weeks ago...
The two esters should have different scents.
I found only one in a public database:
ethyl phenoxyacetate
green floral rose
I did not find the opposite version which is not available in any public scent database, just the closest one which is isobutyric acid ester of your
alcohol instead of acetic acid
phenoxyethyl isobutyrate
sweet fruity tropical rose honey floral waxy
fruity sweet green apple powdery floral
Boffis I hope such dark era of organic chemistry will never happen again and we attract more chemists to the beauty we are touching.
|
|
|
aromaticfanatic
Hazard to Others
Posts: 173
Registered: 10-9-2019
Member Is Offline
|
|
I'm not too familiar with how this site works but wouldn't this be great to add to prepublication?
|
|
|
|









