night429
Harmless

Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood: 
|
|
Synthesis of 4-chloro-2-nitrophenol
I followed the procedure outlined on page 109 of "Fundemental Processes of Dye Chemistry" scaled down, and with two major changes to the reaction
conditions—instead of using an autoclave at 3atm and 145°C, I just did the reaction at atmospheric pressure and at around 100°C.
I put 3.0g of sodium hydroxide in roundbottom flask and added to it 65.5mL of water. Once it had all dissolved, I added 4.8g of
2,5-dichloro-nitrobenzene, which stayed as a solid, and then some sand to help it boil evenly. I then attached a water-cooled condenser to the flask,
put it in a sand bath, and heated up.
I kept it at a reflux for 15 hours. As it heated up, the 2,5-dichloro-nitrobenzene melted and formed a yellow layer at the bottom of the flask. The
aqueous layer gradually took on a yellow color, then a reddish, over the course of the first 4 or 5 hours. I left it overnight, and in the morning,
the color had gotten slightly more red. There was still a layer on the bottom of the flask, which caused the flask to bump occasionally.
At the end of the 15 hours, the flask looked the same as it did that morning. I took it out of the sand bath and let it cool to room temperature, then
put it in the snow to cool it as much as possible. Once it had cooled to below room temperature, the 2,5-dichloro-nitrobenzene had solidified at the
bottom of the flask. I filtered the solution through a coffee filter to try to catch any product that the procedure had said would form. I ended up
only getting some sand and a small amount of 2,5-dichloro-nitrobenzene (nothing was the red color of the product).

Solution cooling down
I slowly added some 31% hydrochloric acid to the filtrate, which caused the color to change from a reddish to a pale yellow, and precipitate formed. I
then filtered this through some coffee filters, washed it with a small amount of water, then dried it.

Final product
I haven't had the time to properly test its properties (namely melting point), but just because of the color of both the basic and acidic solutions,
and the fact it was pH sensitive, I believe that the material I recovered is 4-chloro-2-nitrophenol. I only recovered 0.186g of it, which corresponds
to a yield of 4.3%. I attribute this to both a lack of stirring, which would have dispersed the 2,5-dichloro-nitrobenzene better, and the fact that I
was doing the reaction at a significantly lower temperature than the procedure called for.
I did recover most of the 2,5-dichloro-nitrobenzene since it was a separate, solidified layer. I filtered it off, then washed it with water a few
times. It weighed 4.5g, which means only around 0.3g of it reacted. I then recrystallized from isopropyl alcohol it to make sure it wasn't
contaminated with sand or other impurities and put it back with the rest of my 2,5-dichloro-nitrobenzene.
"Let's spin apart while racing through the atmosphere"
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Nice bit of work night429. I had been looking at this route to 2-nitro-4-chlorophenol-6-sulphonic acid since I now have access to large amounts of the
precursor but the requirement according to Fundamental Processes of Dye Chemistry for vigorous stirring in an autoclave had put me off. I was thinking
of trying a process similar to yours but using an excess of much stronger NaOH solution. My reasoning is that 40% NaOH and stronger solutions have a
much higher boiling point than say 5-10% and hopefully this would force the reaction along nicely. The reason for the autoclave is that it allows
industrial producers to drive the reaction to completion with a very small excess of NaOH and at the same time avoid loss of the volatile
nitrochlorobenzene. In the end I decided to go via urea fusion to 2-nitro-4-chloroaniline and then decompose the diazonium salt to the desired
2-nitro-4-chlorophenol. I haven't tried this yet because I decided to try the phenol -> 4-chlorophenol -> 4-chlorophenol-sulphonic acid and the
nitration to the target compound. This process took a bit of experimentation but I have now got it to work well. The main issue being the separation
of the chlorophenol isomers.
Your route would be a cleaner and shorter route if you can get it to work. Keep up the good work! By the way did you make your own
2,5-dichloronitrobenzene from p-dichlorobenzene?
I have just looked again at Fierz-David's book and they use 40deg Beaume sodium hydroxide this is 35% NaOH W/V.
[Edited on 16-2-2021 by Boffis]
|
|
|
night429
Harmless
Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood:
|
|
Thanks Boffis! I was hesitant to use more sodium hydroxide as the book warns that too concentrated sodium hydroxide can cause "resinification", and
yields are "appreciably lowered". I was under the impression that the 40° Beaume solution was added to an additional 600mL of water in the reaction
flask.
Nonetheless, I'll see if a higher NaOH concentration improves yields, especially since the temperature isn't as high. I'm also considering using a
mixed solvent like water/glycerin to increase the boiling point. I'll continue to update this thread once I get a chance to redo it.
And yes, I did make the 2,5-dichloronitrobenzene from p-dichlorobenzene, following the procedure you posted on a 1/4 scale! Although, I modified it
slightly: using manual stirring and replacing sodium nitrate with an equimolar amount of potassium nitrate. It seems to have worked well, and I got a
77% yield after recrystallizing from isopropyl alcohol.
"Let's spin apart while racing through the atmosphere"
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I was not so worried about resinification or lower yields because of the lower temperatures that I would get with just boiling sodium hydroxide. The
use of a higher boil point solvent is a good idea but I would go for something like glycol rather than glycerol as the later does tend to decompose on
heating with salts
I'm glad to see someone has benefited from my work on p-dichlorobenzene!
By the way do you have a target beyond the 4-chloro-2-nitrophenol?
|
|
|
night429
Harmless
Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood:
|
|
That's a good point. The last thing I'd want is to deal with something like acrolein.
My target is make some azo dyes with the derivatives and document them, since the azo dyes produced aren't well documented to my knowledge (aside from
the vivid blue one with chromotropic acid). The derivatives I hope to make are:
2,5-dichloroaniline (I know you synthesized and steam distilled this)
4-chloro-2-aminophenol
2-amino-4-chloro-6-nitrophenol
Potentially 4-chloro-2-aminophenol-6-sulfonic acid
I know the latter can be used in interesting chromium complexes, but the synthesis of the other reagents needed for those seems rather difficult.
As for the coupling reagent, I plan on making 1-naphthol, since I don't have too many other coupling reagents, and 2-naphthol seems like it needs
extended (14 hours) heating at 160°C with no reflux—which something I can't do in this weather. Do you think that hydroquinone could potentially be
used as one? If so, since the ortho position is the only position available, I wonder if there would be some kind of azo/hydroazo tautomerization that
would stabilize the molecule, like with Sudan I and some other 2-napthol-based dyes.
"Let's spin apart while racing through the atmosphere"
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Its difficult to couple quinol and catechol with diazonium salts as they tend to act as reducing agents towards the diazonium salt. Catechol azo dyes
have been prepared but as far as I know only by indirect routes. I have a paper somewhere on preparing these dyes by coupling the diazotised amine
with guaicol (o-methoxyphenol) and then de-methylating the ether but I have never tried it. I wonder if the same effect could be obtained from the
quinol or catechol monoacetate esters? These would be easier to cleave afterwards I think. Salicylic acid is a good OTC coupler and there are a fair
number of heterocyclic compounds too (pyrazolones, barbituric acid and imidazoles for instance) and the resulting dyes have strong complex forming
abilities.
I have prepared 4-chloro-2-aminophenol-6-sulphonic recently too. The work I posted is mostly to do with the preparation and separation of the
chlorophenol-sulphonic acid isomers, these nitrate very easily, so much so that its difficult to stop the nitration at the mono stage with the
p-chloro acid as nitration tend to displace the sulphonic acid group too. I am currently working on scaling up the procedure, the tricky bit is the
nitration of the sodium 4-chlorophenol sulphonate without getting 2,6-dinitro-4-chlorophenol.
|
|
|
night429
Harmless
Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood:
|
|
Ah okay, that makes sense. I could try to prepare and use the monoacetate ester when I get the chance and see if they'd work, although I'll use
p-aminophenol to test it, since it's cheap and easy to prepare.
Salicylic acid is a good idea, and I'll definitely try that, too, especially since the resulting dye should theoretically be able to form salts.
Yeah, the partial nitration definitely seems like a tricky thing to control, and I wish you the best of luck in scaling it up!
"Let's spin apart while racing through the atmosphere"
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi night429, did you try this experiment with a glycol solvent?
I have a 0.1 Molar scale experiment running at present with ethylene glycol as solvent and it seems to be working, Its been simmering at about 125-130
C for the last 3 hours. The temperature is a bit approximate as I can only dip the bottom 15mm of the thermometer into the mixture and its a total
immersion type. The biggest problem I have encountered so far is the condensation of the vapours to prevent lost of nitrocompound. I am using a RB
flask filled with warm water as a condenser (cold water causes the nitro compound to condense as a solid and not run back into the beaker). When I
last tested the temperature the liquid on the thermometer bulb was a beautiful garnet red so something is happening.
|
|
|
night429
Harmless
Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood:
|
|
Hi Boffis, I haven't had the opportunity to run the experiment yet. It definitely sounds like something is happening, and the red color is a good
sign! For whatever reason, I didn't even consider that the vapors of the nitro compound solidifying would have been an issue when not using water as a
solvent.
Let me know how the rest of the reaction goes!
"Let's spin apart while racing through the atmosphere"
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Badly 
I am now trying to clean up the crap.
After 10 hours I let the mixture cool and this morning found a viscous mass. I diluted it with 150ml of water and because there was so much unreacted
dichloronitro compound present I added 50ml of DCM to get it into solution. I then filtered the whole lot hoping to get some sodium
nitirochlorophenolate let on the filter but all I got was a little gloopy red brown tar. The DCM-aqueous mixture wouldn't separate so I acidified it
with HCl. That did the trick but I had to filter off more tar. This filtrate separated nicely and I now have about 50ml of deep red extract but I
don't hold out much hope. I will remove the DCM and then probably steam distill it. This should give me a tar-free mixture of nitrochlorophenol and
dichloronitrobenzene which can then be separated with NaOH again.
All in all a very troublesome prep and a messy clean up. Definitely not one of my more successful experiments!! I think the direct nitration of
4-chlorophenol may be a better route for me as I have this compound.
I am going to try the conversion of 2,5-dichloronitrobenzene into 2-nitro-4-chloroaniline by the urea fusion method and I may also try the aqueous
ammonia or ammonium carbonate in an autoclave route too. I will then try to convert the aniline into the phenol via the diazonium salt.
I have tried the the urea fusion route with several nitro-halobenzenes and found that p-nitrobromobenzene and 2,4-dinitrochlorobenzene work well but
2-nitrobromobenzene gave a very small yield. So the method works for sufficiently reactive nitro-halobenzenes.
[Edited on 23-2-2021 by Boffis]
|
|
|
night429
Harmless
Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood:
|
|
Sad to hear that it tarred up! That's a good idea, and I'm interested in hearing how the diazonation goes.
I've just redone my original synthesis, albeit at half the time (8hr instead of over 15). I also used four times the amount of NaOH. I'm currently
waiting on a yield, but I did get some nice red needle-like crystals that had formed upon cooling.
Red crystals of the sodium salt after filtration
Acidification of the filtrate gave a small amount of pale yellow crystals. I'm currently waiting on them to dry, but the yield seems to be poor like
last time. I'll keep you updated on how it goes.
"Let's spin apart while racing through the atmosphere"
|
|
|
night429
Harmless
Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood:
|
|
An update to my previous post:
The yellow crystals dried to an off-white, which concerned me, since the previous 4-chloro-2-nitrophenol was a yellowish. I'm unsure as to why this
happened, but nonetheless the amount of it recovered was minimal (I presume that the majority of it precipitated as the sodium salt due to the amount
of sodium ions present in solution).
The sodium salt dried to a nice crystalline powder. I didn't get the chance to mass it, but the yield is poor just by looking at it.

Dark red crystals of the sodium 4-chloro-2-nitrophenolate
Another procedure I did involved an attempted reduction of the nitro group in 2,5-dichloronitrobenzene using polysulfides and isopropyl alcohol to
help dissolve the starting material. I used 1.5g of the 2,5-dichloronitrobenzene, 0.3g of 90% sulfur dust, and 0.5g of sodium hydroxide. I boiled the
sulfur and sodium hydroxide together in around 20mL of water, and heated the 2,5-dichloronitrobenzene in a mixture of 23mL 91% isopropanol and 30mL of
water, which yielded a very slightly cloudy solution. The color of the solution turned to a dark brown after adding the orange/brown polysulfide
solution.
I filtered off the solids, manually pressed out almost all of the solution into some paper towels, and added the filter cake to around 40mL of
heptane/ether mixture. This was heated until boiling, then poured into a ziplock bag to separate off the organic layer from the trace amount of water
(as well as some inert impurities left over from the impure sulfur). I poured the yellow organic layer into a test tube, where it cooled down and a
small amount of needle-like crystals had formed. I filtered these off and allowed the solution to evaporate on a watchglass. I recovered around 1.0g
of a yellow crystalline mass, and the white needle-like crystals that were filtered off were discarded.

Bad picture of the recovered crystalline mass
As I don't have TLC plates, I tested some other things, including pH sensitivity, an azo dye synthesis, burning it from below, and smell (melting
point can't be used, since the references I've seen say the melting point of the product is extremely close to the melting point of the starting
material)
The pH sensitivity test showed it only slightly dissolved in any concentration of HCl, if at all, giving a yellow color.
The azo dye synthesis only resulted in a very slight orange color upon addition of salicylic acid, but no precipitate formed.
Burn test compared to the starting material showed it was more prone to burning with a flame instead of just evaporating, but this isn't
substantial enough.
The smell was more acrid than that of the starting material, which smelled somewhere between p-dichlorobenzene and nitrotoluenes.
Due to these tests, I don't believe that the product is pure 2,5-dichloroaniline. But, I don't think the product is just starting materials. The
reason I say this is because the product is a bright yellow, but the 2,5-dichloronitrobenzene that I started with is an off-white (very similar to the
color of sodium nitrite). So, I'm quite honestly unsure what I have. Maybe it is just the starting material that's yellow from a different solvent of
recrystallizing? Since I'm back at university, I'll be unable to test any hypotheses I think of for the next two weeks.
"Let's spin apart while racing through the atmosphere"
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Interesting work night429. I used the iron filings+HCl method and then steam distilled out the aniline. This works well, dichloroaniline is very
volatile and has a characteristic smell that is similar to aniline + p-dichlorobenzene to me and it sublimes spontaneously so that the solid becomes
covered with fury needles on standing in a closed container at warm room temperatures (>20 C). It forms a hydrochloride but it is not very soluble
in conc HCl but can be isolated. The hydrochloride can't be recrystallised from water since it hydrolyses though it reportedly can from absolute
ethanol (and presumably therefor from dry IPA too). It is not easily diazotised according to a paper I have found. The paper is in German but I am
currently translating it as it contains a lot of interesting reactions of 2,5-dichloroaniline. I'll post it here when I have finished.
|
|
|
night429
Harmless
Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood:
|
|
Thanks! I was unaware that it wasn't very soluble in hydrochloric acid. I don't have access to my supposed 2,5-dichloroaniline, but considering I'm
storing it at room temperature, if it really is 2,5-dichloroaniline, I should be greeted to the fluffy crystals you're talking about when I next see
it.
Was your product a similar yellow color by the way? I was researching the properties of the 2,5-dichloroaniline and found that the color reported is
varied, a few colors being brown, red, amber, and colorless. I'm looking forward to reading that paper!
"Let's spin apart while racing through the atmosphere"
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
My 2,5-dichloroaniline is a bit mottled. The solidified oil that separates as steam distillation progresses is an amber colour but the crystals that
formed from the aqueous phase on cooling over night are pure white felted needles.
By the way the paper I am translating mentions the coupling with salicylic acid as taking 24 hour to complete!! The beta-naphthol sulphonic acids they
mention couple almost instantly. The paper also states that a 7-8 fold excess of mineral acid is required during diazotisation to suppress the
formation of the corresponding diazoamino compound.
 Steam distilled 2,5-dichloraniline Steam distilled 2,5-dichloraniline
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I've posted the translated paper I referred to above in my thread on derivatizing p-dichlorobenzene:
http://www.sciencemadness.org/talk/viewthread.php?tid=156633...
|
|
|
night429
Harmless
Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood:
|
|
Thanks Boffis, I'll take a look at it! I'm surprised to hear that the reaction time is so much longer than the formation of some of the other dyes.
"Let's spin apart while racing through the atmosphere"
|
|
|
night429
Harmless
Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood:
|
|
So, after a couple of months, I finally decided to revisit this. I don't know what happened, but I got a very different outcome than
the paper.
I made the 2,5-dichloroaniline via the Bechamp reduction of 2g of 2,5-dichloronitrobenzene with steel wool in HCl. I then added NaOH until the
solution turned black, then steam-distilled out the aniline. I recovered 1.1g of white product (66% yield), and it smelled like fresh asphalt.
Fresh 2,5-dichloroaniline:

To make the diazonium salt, I followed the paper scaled for 100mg of 2,5-dichloroaniline, although I improvised just a bit (used more sulfuric acid
because I couldn't measure 0.25mL and used more water overall). When I added the sodium nitrite, a yellow solution occured (as reported) with no
precipitate.
As for the actual azo coupling, I improvised a bit. I measured out a molar equivalent of salicylic acid, added a small amount of water, then added a
bit of NaOH until it dissolved. I cooled it, since it had gotten decently hot, then I added a drop of the diazonium solution. Much to my surprise,
there was an instant reaction, and a vibrant orange color formed. I kept adding the diazonium solution, and eventually the color turned to a muddy
brown. I thought that there was some kind of rapid decomposition occuring, so I added a pinch of salicylic acid. When that did nothing, I just added
the rest of the diazonium solution all at once.
The solution still was a brown, and then I realized it must have been a pH thing, so I tested it. It came out to very acidic, so I just added an NaOH
solution, and the color changed back to the vibrant orange. Now that I realized the dye was simply pH sensitive, I got more HCl to precipitate out the
dye, then filtered it off.
Acidic solution:

Basic solution:

The yield I got was 174mg of a dry, rich-brown powder, which is a surprisingly-high 90.6% yield (This dry powder, when added to a basic solution,
dissolves and forms the orange color as seen before):

I have no idea why my final product is so different than the one reported by the paper. The formation of the dye was instant, and the color was
strikingly different than the "light-yellow" color reported. Could I have done something wrong? Or, is the paper the one that's wrong?
"Let's spin apart while racing through the atmosphere"
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi night429, nice work, its always good to see some practical chemistry being done. The colours referred to in the paper are the colours of the dyed
wool and not necessarily the colour in solution. I presume these colours are the ones without mordant. That you found it reacts so quickly is strange,
these old German papers are usually pretty accurate in their observations.
Being a salicylic azo dye it should form insoluble lakes with Al, Sn, Fe, Cr etc and you may be able to mimick the colours by spotting on filter paper
and then adding a drop of the metal salt solution.
I still haven't found a good way to convert the dichloronitrobenzene into the corresponding phenol directly but when I get home I am going to try
converting it into the aniline and then to phenol via diazotization.
|
|
|
night429
Harmless
Posts: 45
Registered: 12-11-2019
Member Is Offline
Mood:
|
|
Thanks Boffis! That makes sense, and I know practically nothing about actual dye processes. I tested the lake properties with a few different metals I
had on hand: iron(ii), iron(iii), and zinc(ii). The way I did it was by putting a drop of dye solution (I added a few granules of NaOH to get it to
dissolve) onto a white surface, and then adding a drop of metal solution. Once I did that, I scaled it up in containers so I could recover some amount
of the product. The results are as follows:
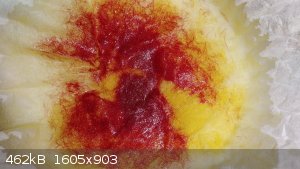
Both iron(ii) and iron(iii) gave a green color (this is the iron(ii)):

Zinc(ii) gave a pale yellow:
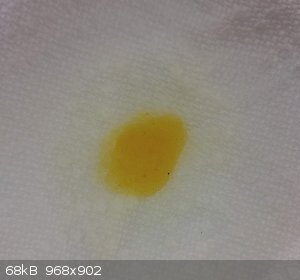
Looking forward to hearing about the phenol synthesis!
"Let's spin apart while racing through the atmosphere"
|
|
|








