Fery
National Hazard

Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
pharma / food grade ethanol - distillation using Hempel column
In spring 2020 I collected 10 kg apples which fall to the ground in an abandoned apple orchard. I processed them using blender, added 7 kg beetroot
sugar and water so the volume was 40 L and let it to ferment. When fermentation finished and CO2 evolution stopped, I divided the cider into 3
portions and distilled it using my 20 L stainless steel kettle which I use also for mentha piperita hydrodistillation. I did not made photos as there
is nothing special, the photo of the apparatus is in the thread with hydrodistillation. I performed the distillation during winter while burning wood
in my stove to heat the house, so it did not cost extra energies like electricity / gas. I got 3 portions of 3 L so together approximately 9 L of
35-40 vol % product (the apparatus has some very simple and not too much efficient column/head. Then I distilled it at once for the second time using
the same pot, it yielded cca 4,5 L 75-80 vol % concentration. Again nothing special.
Now it was the right time to perform final distillation. As I have 6 L RBF it would fit into it but my largest heating mantle is designed for 4 L
flask. So I had to divide it into halves and perform the final distillation twice. I did not want to distill on wood stove as the heating power is not
steady. I did not want to distill on gas stove because of the risk of fire if the flask breaks (well, I could use big pot with oil bath but I do not
have such a big pot and there is also not enough room above my gas stove upto the ceiling, the tallness of the whole apparatus is 1,8 m).
So I put the 75-80 % alcohol into 4 L RBF, then assembled 1 m high 30 mm inner diameter Hempel column packed with 6-10 mm Raschig rings, head for
taking off reflux at desired ratio, collecting flask. I greased the stopcock with as little as possible of food grade grease Lukosan M 14. I insulated
the column by approx 10 layers crumpled newspaper (keeps more air which is the insulator when compared with flat newspaper) and finally aluminium foil
layer so it was more airtight (newspaper is not too much airtight). Well it does not compare to vacuum jacketed column, but I do not have such
insulated column. The unjacketed column was not much expensive, IIRC it cost only somewhat like 20 EUR.
The main fraction distilled at 77,25-77,5 C (lit. 78,2 C azeotrope) and water distilled at 99,5 C (lower air pressure, overcasted, little rain).
Photos from the process follow and also short video here
https://youtu.be/0wMHckg64iA
on the 1st picture the flask with attached column, 2nd detail of packing with Raschig rings which were wetted with the distilled liquid by pouring the
liquid into the apparatus from the top of the column, 3rd on the flask bottom there are red/brown boiling stones and the flask is already filled with
the liquid to be distilled
  
1st column insulated with approx 10 layers of crumpled newspaper, 2nd final aluminium foil to at least somewhat increase airtightness, 3rd fully
assembled and filled apparatus
  
1st also the flask was insulated with newspaper, 2nd condensate already dropping in the regulated reflux ratio distillation head, 3rd the T was
initially high as the column was wetted with the liquid to be distilled
  
1st column flooding at which the heat power was reduced so the column later did not flood and the speed of reflux returning was as high as possible
(the higher reflux amount returned into the column, the better separation during distillation, but the column must not flood), 2nd T falling slowly,
3rd column not flooding, stopcock still closed
  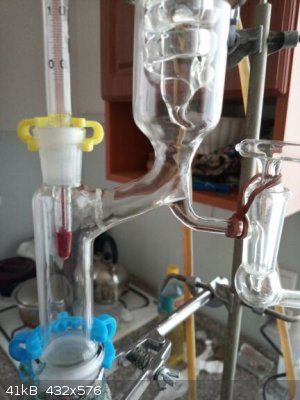
1st minimal T reached, T 74,0 C and does not fall anymore, after 30 minutes the equilibrium in column already established, 2nd and 3rd the stopcock
slightly opened, so approx 1 drop taken into the collecting flask and 9 drops returning back into the column
  
1st forerun collected in a small 100 ml flask, 2nd T raising, actually 75,25 C, 3rd T rasing, actually 76 C
  
1st and 2nd drops of condensate collected in small flask (forerun), 3rd T raising, actually 76,5 C
  
1st T 77,0 C, 2nd still collecting forerun, T reached 77,25 C at which it did not raise anymore, I collected 100 ml of extra forerun at this T to
certainly remove all methanol during which the T kept stable at 77,25 C, 3rd switched the collecting flask from small 100 ml into 1 L RBF, the main
fraction distilled in T range 77,25-77,5 C (low atmospheric pressure caused by weather conditions - overcast, occasional slight rain)
  
more photos will follow, I had to split due to limitation of number of photos in 1 post
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
here how quick the T changed when the main fraction distilled, every picture name contains its exact date and time
now collecting afterrun and finally determining the boiling point of pure water as the atmospheric pressure was below normal (caused by weather
conditions)
the main fraction distilled at 77,25-77,5 C, I managed to capture the quick T raising with the afterrun fraction
17:07:09 T 78,0 C, 17:07:29 T 78,25 C, 17:07:43 T 78,5 C
  
17:08:03 T 78,75 C, 17:08:12 T 79,0 C, 17:08:26 T 79,5 C
  
17:08:39 T 80,0 C, 17:08:47 T 80,25 C, 17:08:53 T 80,5 C
  
17:08:58 T 80,75 C, 17:09:02 T 81,0 C, 17:20:32 T 99,5 C water distilling after collecting the whole afterrun, T does not raise anymore, low
atmospheric pressure caused by weather conditions
  
|
|
|
unionised
International Hazard
Posts: 5102
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
Could you include a few more pictures of a thermometer reading nearly 80C?
:-)
|
|
|
Sulaiman
International Hazard
Posts: 3554
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Offline
|
|
nice column, very nice take-off head !
I don't trust alcohol thermometers long term and
maybe immersing the thermometer more would give a more accurate reading ?
(or was it just local atmospheric pressure ?)
CAUTION : Hobby Chemist, not Professional or even Amateur
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
I got better idea for thermal insulation of the column. In every shop supplying plumbers there are thermal insulation tubes (plastic material with a
lot of small air bubbles) of various internal diameters and wall thickness, they are used to insulate water tubes etc. I'm not sure whether they
withstand 100 C at the end of distillation. So maybe cover the glass column with few layers of newspaper + aluminium foil (in case plastic insulation
melts) and then cover that with insulation tube from plumbing shop. They are 2 m long which is much more than my longest distillation column.
Sulaiman - it was local atmospheric pressure. I'm living at altitude 430 m above sea level and that day the air pressure was lower due to weather
conditions. I checked that water boils at 99,6 C when atm. pressure 99,8 kPa instead of standard 100,0 C at 101,3 kPa.
For ethanol the boiling point change could be also calculated (IIRC Clausius-Clapeyron equation). But even without calculation, the T stayed in range
narrower than 0,25 C for the whole main fraction. You do not need to calculate the T difference to watch when T reaches it, by my experience it is
enough to watch the thermometer without knowing the exact b.p. I was distilling 2 liters so when T does not increase (I was able to determine T change
0,25 C by my thermometer) during collecting 50 ml of distillate you are at the right temperature and you should collect the main fraction. Removing
extra 50 ml from the beginning has negligible impact on yield at scale 2 L and is also good to remove as much methanol as possible. I will send
samples to my friend Bedlasky who will determine methanol content, he has access to gas chromatography. Boiling point of the main fraction +- 1 C may
be caused by the altitude and weather changing atmospheric pressure. I distilled the second half the next day and at the end of distillation water
distilled exactly at 100,0 C (better weather and normal atmospheric pressure that day). I won't post pictures of the second day distillation when T
was around 80 C and later 100,0 C according unionised suggestion.
There is a norm for thermometer producers in my country so they must write to the thermometer stem a value how much millimeters the thermometer bulb
should be inserted for thermometers without ground glass joint. All heads and distillation adapters which are made in my country are for 40 mm
insertion length of thermometers with ground glass joint (40 mm is the length of thermometer bulb and stem measured without the length of ground glass
joint 14/23) - all these glassware pieces are compatible with these 40 mm ground glass joint 14/23 thermometers. Also the 10th and 12th picture in my
initial post show the position of thermometer bulb which is ideal.
|
|
|
Fyndium
International Hazard
Posts: 1192
Registered: 12-7-2020
Location: Not in USA
Member Is Offline
|
|
I have used those tube foam insulator wraps and they are great.
The concept of that still is same as bokakob. It works great.
|
|
|
Sulaiman
International Hazard
Posts: 3554
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Offline
|
|
I'm used to 75mm (3") immersion depth which makes 40mm look wrong to me,
but 40mm is more practical for small scale glassware than 75mm.
At home (UK) I use this stuff https://www.paroc.co.uk/products/technical-insulations/pipe-...
but here I've been using microfiber cloth + Al foil for column insulation.
CAUTION : Hobby Chemist, not Professional or even Amateur
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
My friend Bedlasky performed GC analysis and methanol was below 0,1%, higher alcohols below 0,03%, ethyl acetate below 0,005% in every sample (I sent
him 3 samples from the 2 distillations, both distillations yielded approx. 2x1,4 L of ethanol, first 1 L stored in a 1 L flask - sample 1., sample 2.
and last approx 2x0,4 liters merged and stored in the third 1 L flask - sample 3).
I was very afraid of methanol as I used 10 kg apples fallen to the ground/grass as a source of yeast + minerals + organic substances for yeast. To
increase yield of ethanol I used sale-out sugar from supermarket. Apples are reach in pectins and fermentation of methylated pectins yields methanol.
I rather let this ethanol for my future pharma / food purposes (like tinctures etc) and eventually purify petrochemical ethanol for my syntheses
(remove denaturing agents MEK and bitrex).
|
|
|
Fyndium
International Hazard
Posts: 1192
Registered: 12-7-2020
Location: Not in USA
Member Is Offline
|
|
Homedistiller has investigated this thoroughly, and the methanol is trivial to separate from ethanol even with smaller reflux fractionating column.
I have concluded that there are no known cases of methanol poisonings from fermentation based moonshines. They all derive from added technical
alcohols to cut costs.
I do indeed suggest using otc ethanol sources for synthesis. It's significantly cheaper and can be easily purified, depending on purpose, from
technical to even into potable form. Fermentation is extremely long and arduous process, and really works well only in larger scale. For drinking
purposes, 50L boilers tend to be minimum, as single strip&frac can deliver only about 3-4L of azeotropic high quality potable alcohol. Considering
that you need vats to ferment it for at least a week + few days for clarification, then strip it to 50% and then fractionate over several hours to
recover the foreshots, heads, hearts and tails. Of these, you recover about 50-60% at max if you want the high quality only without off taste. Even
then, sitting on activated carbon for couple of weeks at 50% dilution will improve the profile noticeably.
Instead, you go to shop, buy 10L canister of ethanol for 20€ and simple distill it at full rate to recover almost 9 liters of azeotropic ethanol
with little MEK in few hours. Add some NaOH, and the MEK is also gone. Distilling it twice and sitting on carbon has made it potable to some of the
more sippy chemists.
[Edited on 1-5-2021 by Fyndium]
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Fyndium 1 L tech. ethanol costed here 50 CZK before virus, then producers/sellers/resellers abused the situation so the price skyrocketed to 3 fold
and not only price but it was also unavailable together with isopropanol. There are still some sellers which sell it at such high price although no
such demand for disinfectants and ethanol and isopropanol already returned to the lists of eshops.
https://www.funchem.cz/www-funchem-cz/eshop/2-1-ORGANICKE-LA...
1 L for 149 CZK, still threefold price. Maybe they stocked a lot of it it 1 year ago while the prices were too high so they have to sell it so
overpriced.
I can produce 1 L of ethanol from 2 kgs of sugar which I can buy for 2x7,90 CZK = 16 CZK in supermarket sales-out which is 3-times cheaper than the
price of synthetic ethanol before SARS-CoV-2 (50 CZK). You can approximate 50 CZK = 2 EUR = 2,30 US$. During winter when heating my house by burning
wood in a stove it does not cost extra energy, just putting the still to the stove. So processing the large volumes of diluted ethanol requires only
some work and does not cost me any money. Only the final column distillation does require electrical or gas heating as the wood heating is not steady.
Moreover in my ethanol there is no shit like 1,6 % methylethylketone either denatonium benzoate.
For removing MEK using NaOH you need an old flask for which you do not bother etching glass by hot NaOH. I certainly wouldn't use new shiny glass
flask for that.
Even homemade distillates which may contain upto 1% of methanol (you can't simply distill on column as that would also strip off not only methanol but
also all aroma) do not cause acute methanol poisoning as the poisonous substances are formaldehyde and formic acid which are made from methanol by
liver alcohol dehydrogenase which is competitively processing ethanol in large excess.
I also wanted low methanol content for the case of esterification.
According technical ethanol I was thinking of fuel E85 which you can buy at some petrol stations - the ethanol content is 85% and the 15% is gasoline,
the cost of this fuel is approximately 1 US$ for 1 L. It would avoid etching flask with NaOH as there is no MEK, moreover the alkanes content would
remove all water as azeotrope during distillation, so the obtained ethanol would be close to anhydrous.
Maybe my 1 m long 30 mm diameter column packed with small Raschig rings would be sufficient to separate azeotr. ethanol from MEK (b.p. 78,2 C vs 79,6
C) but such distillation would require certainly something like at least 20 theoretical plates so only then I won't need to use NaOH for MEK aldol
condensation?
|
|
|
Bedlasky
International Hazard
Posts: 1219
Registered: 15-4-2019
Location: Period 5, group 6
Member Is Offline
Mood: Volatile
|
|
Fery: Very nice work!
1l bottle of ethanol from Severochema cost somewhere between 75-85 CZK, depending on the shop. 149 CZK is heavily overpriced.
[Edited on 1-5-2021 by Bedlasky]
|
|
|
Mateo_swe
National Hazard
Posts: 501
Registered: 24-8-2019
Location: Within EU
Member Is Offline
|
|
Excellent results, nicely done.
Time to buy some sugar, turbo-yeast and fire up the old distiller.
I need some ethanol for chemistry experiments and distilling store bought drinking ethanol is very expensive.
One thing i want to comment on.
Why did you not dilute the distillate from first distillation down to 40% and run it through active carbon of good quality?
Then re-distill following your procedure.
Dilution to 40% is because the carbon absorbs better at 40% compared to stronger %.
This active carbon cleaning procedure is done always when making drinking ethanol and its essential for getting rid of foul smelling and tasting
impurities.
And the better coal the better ethanol, active hi-quality stone carbon from brewer stores give best ethanol.
For drinking that is, but i see no downside to do it except maybe the added cost.
It must be a good step when making high quality ethanol for experiments too.
Seeing you get 99.9% purity without carbon treatment makes me think it might not be necessary but i think i try it with carbon.
How did you get absolute ethanol, or did you stay at the azotrope?
[Edited on 2023-1-17 by Mateo_swe]
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi Mateo_swe. Diluting concentrated alcohol is waste of energy and vanished 1 distillation. If you need 40% alcohol then you can apply active charcoal
after the first distillation which concentrated 10% alcohol to 40%. The second distillation concentrated alcohol from 40 to 80% so smaller flask could
be used for the final column distillation step.
I do not plane to drink this alcohol so I did not apply active charcoal. I only needed ethanol without nasty methyl ethyl ketone which is in my
country in every denatured alcohol. So I try to use methanol and isopropanol everywhere possible which are sold in pure form and they both are also
anhydrous. Anhydrous ethanol is again too overpriced here and preparing it by myself requires extra time and effort. So I let the ethanol in azeotrope
concentration. For esterifications I always use Dean-Stark trap with entrainer (isohexanes from hydrogenated medicinal petrolether, another good
choice is to use cyclohexane) to remove reaction water, which also saves time as I see that the reaction is complete, it also increases yield.
I used to distill various fruit spirits since I was 14 years old while I was still in the elementary school. Both my grandfathers liked to dring
alcohol unlike me. I liked the process of producing it but I did not like to drink it. I liked the chemistry behind it and beautiful scents these
fruit spirits have. I used to distill fermented plums, apples, cherries, sour cherries, pears,... I planted a lot of various sorbus species (like
sorbus domestica, sorbus torminalis), wild cherries, junipers, medlars, cornus mas, mulberries into my forest into its south part, which has a border
with agricultural field so there is enough sun, just to try to ferment them in the future (if not me, then someone else will continue).
I always did everything by myself since the beginning (fermentation as well distillation) because I had no one to give me qualified advices. The
quality improved with time and skills. I realized the fruits have to be clean, I used to pick them from trees instead from ground (dirt/soil
introduces unwanted bacteria and you want only saccharomyces which are fungi). I realized slow fermentation is better than fast, so the temperature
should be lower, but not too much low to stop the fermentation. Too high temperature degraded quality so the process of fermentation had to be slow
too as it produces heat. Crushing plums at the beginning caused faster fermentation so later I did not crush them, but put them whole inside the
fermenting vat and used long wooden stick to mix the content after few weeks for the first time (but this cannot be applied for apples where you need
to crush them before filling the vat, but could be applied for very soft variety of pears). Also filling vat not at once but continually as more fruit
ripened was better as the process of fermentation was slower (e.g. some fruits which ripe during 1-2 months, first parts were already partially
fermented when adding following crops of fruits which desirably slowed down the speed of fermentation). At elevated temperature also vapor pressure of
aromas in the vat increases so the loses of aroma into escaping CO2 which is quite big volume of gas. The lower temperature the more aroma kept and
better quality.
The same for distillation, slower = better. Higher speed = more drops carried with vapor into condenser. Higher speed = risk of burning organic
compounds on bottom of still during first distillation when the mass is quite thick.
Turbo yeast you plane to use produce side reactions so you will need the carbon cleaning procedure. In the experiment I posted I used apples from an
abandoned apple trees orchard (no pesticides, no artificial fertilizers). Your turbo yeast will ferment sugars very quickly in few days at elevated
temperature - maybe some cooling could improve quality by reducing side reactions at the cost of waiting few more days longer?
The column distillation got rid of the apple scent completely so if you want beverage alcohol you need simple distillation or less effective column
with less theoretical plates. Btw I collected the forerun from column distillation and kept it as I like its apple scents, in 100-200 ml there is
concentrated aroma from few dozens kg of apples... Also whole ethyl acetate as it distills as an azeotrope with ethanol at lower temperature (in fact,
not binary, but ternary ethyl acetate + ethanol + water).
Also adding sugar from sugar beet is not good for beverage alcohol as it introduces scent of sugar beet and dilutes the aroma of fruit (more ethanol
produced for the same amount of fruit). But as I wanted to produce ethanol for chemistry (methyl ethyl ketone free) I did not worry and added sugar
from sugar beet.
[Edited on 18-1-2023 by Fery]
|
|
|
Mateo_swe
National Hazard
Posts: 501
Registered: 24-8-2019
Location: Within EU
Member Is Offline
|
|
Slower fermentation at a slightly lower temp probably reduce impurities as you say, and slower distillation garanteed improves the ethanol.
Running the distillation too hot and fast reduces quality for sure.
I have a stainless steel distiller that takes 25L per go and it have a maybe 8-10cm thick, over 1 meter ss column filled with ceramic rings.
It works very well and has electric heating but i havent used it for many years now but its stainless so it last a lifetime.
I will try make some ethanol for chemistry use.
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Btw this year I fermented 30 kg of strawberries with 4 kg of sugar and produced strawberry distillate for the 1st time in my life (I mean
strawberries). That's because this year harvest was about 100 kg and I did not know what to do with it and finally and luckily I got the idea of
producing a distillate. Strawberries are harvested here in the summer (like 1st June - 10th July) and I performed first distillation during summer
after 1 month of fermentation. I used a yeast from a shop to be sure to have pure fermentation (I used yeast for distillers, not any yeast for beer
and not at all any yeast for kitchen). Such valuable fruit (so much time consuming to harvest them!) cannot be fermented by wild yeast. I processed
the strawberries by washing with plenty of water on a mesh, then I used a mixer to make a smoothie, added sugar and revived yeast (they were in a dry
form in sealed small bags so I had to prepare them 3 days before). Luckily there were here few very cold days this year summer and I managed to do the
1st distillation after 1 month (the fermentation was done in about 2 weeks). The whole house was terribly overheated as I distilled on a stove for
firewood although outside temperature was slightly below 10 C (unusual for a summer but lucky me) and all the windows wide open. A firewood stove is
more gentle for heating (avoids burning the bottom) than using gas flames stove or electrical heating. A firewood stove is also very inefficient and a
lot of heat was transferred into the house instead into the boiling tank.
This is how does it look like, that time I was hydrodistilling mentha herb instead, but the stove and boiling pot are the same:
https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
My friend teodor was here just this time, sorry bro for the heat. The next visit I give you a bottle of strawberry distillate.
Once I got 1st distillate with concentration about 40 % it is stable and I did not have to hurry into second distillation. But the fermented
strawberry smoothie is very sensitive and I had to process it soon after the fermentation was done.
The second distillation was performed during winter when I regularly burn firewood fore heating the house. The final distillate after the second
distillation has amazing aroma. I do not drink alcohol, so I'll give it as gifts to my family. But I like the scent and the process of producing that
and also that there are barely any people producing true strawberry distillate. You can buy here only a fake commercial product which is a pure
ethanol mixed with artificial strawberry aroma or at best a product made by a distilling pure ethanol and passing ethanol vapor through a fresh
strawberry fruits and then into a condenser.
|
|
|
|









