Diachrynic
Hazard to Others

Posts: 219
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Protocatechuic aldehyde from ethylvanillin
Vanillin is notoriously hard to turn into protocatechuic aldehyde. Ethylvanillin, on the other hand, is much easier to dealkylate. While I haven't
found a direct source, german patent DE69507676[5] uses a two step process to convert ethylvanillin into isovanillin, by methylation and
then deethylation. I was curious if ethylvanillin itself could be turned into protocatechuic aldehyde this way.
Alternative names for protocatechuic aldehyde are protocatechualdehyde, 3,4-dihydroxybenzaldehyde or [139-85-5].
Chemicals:
Ethylvanillin
Sulfuric acid (95-97%, pure)
Ethyl acetate
Ice
Petrol ether (for TLC)
Glacial acetic acid (for TLC)
(Sodium sulfate)
Procedure:
In a 150 mL beaker were placed 5.5 g of concentrated sulfuric acid and a stirbar. Then 1 g of ethylvanillin was added and stirred in, the pieces take
on a greenish color before dissolving to a clear yellow solution. The beaker is covered with a watch glass, placed in an oil bath in the fume hood,
the temperature of the oil is raised to 70 °C and maintained for 4 hours. Some smoke or fog is visible in the beaker.
Reaction progress can be monitored by TLC: A glass rod is used to extract a drop of the acid which is dissolved in about 1 mL of water. This solution
is spotted along a reference sample of ethylvanillin dissolved in ethanol onto a TLC plate. Eluent mixture is petrol ether/ethyl acetate 1/1 v/v with
a few drops of glacial acetic acid added. (The Rf values are for 0.2 g per 10 mL eluent.) Staining is accomplished by 254 nm UV (quenching) or by
staining with iodine vapor (more sensitive).
Rf (ethylvanillin) = 0.60
Rf (protocatechuic aldehyde) = 0.37
Samples were taken after 2, 3 and 4 hours. During this time the ethylvanillin spot gets weaker and weaker.
The reaction mixture that has taken on a brown color by now is taken out of the oil bath and left to cool for a moment before it is dumped onto 11 g
of crushed ice while being stirred. A clear solution results. The mixture is almost fully molten but still cold. Then it is extracted once with 10 mL
of ethyl acetate and then twice with 5 mL of ethyl acetate each. The color moves mostly into the organic layer (reddish). The organic layers are
combined, dried over some sodium sulfate and then evaporated to about 2-3 mL volume which is allowed to evaporate at room temperature. Greyish
crystals with a hint of blue and green are obtained and left to dry in air for half a day.
They weigh 0.77 g (93%).
  
The solids are redissolved in about 4.4 g of hot water, giving a brown solution. On cooling to room temperature crystallisation had to be initiated by
scraping with a spatula. The suspension of crystals was then placed for several hours in the fridge. Vacuum filtration over a Büchner funnel provides
a slightly brownish crystalline product.
After air drying it weighs 0.51 g (63%).

TLC (pet ether/ethyl acetate 1/1 v/v + a few drops of glacial acetic acid) shows a large spot at Rf = 0.37 and a faint spot at Rf = 0.60 (small amount
of leftover ethylvanillin).
The melting point was measured to be between 149-151 °C. Ethylvanillin melts around 77 °C, the values for protocatechuic aldehyde range from 150 to
156 °C, depending on the source. Amarego lists 153 °C.[4]
Melting point and TLC thus give convincing evidence that ethylvanillin was successfully dealkylated.
Possible improvements:
The procedure should be carried out in an appropriately sized RBF to avoid the sulfuric acid absorbing moisture from the air. The reaction mixture
should perhaps be stirred for longer. Alternatively, the temperature could probably be increased while the reaction time being decreased. The stirbar
should be submerged because ethylvanillin can stick to it and not react.
The diluted solution could be extracted with more ethyl acetate.
Finally, less water should be used for the recrystallisation, and it could be cooled further in the freezer. It has also been suggested to
recrystallise from toluene.[4]
The patent[5] uses a ratio of 2.7 g of sulfuric acid per gram of 3-ethoxy-4-methoxybenzaldehyde. On this scale I preferred ease of stirring
and used a larger excess of acid, but it might not be required.
Drying the ethyl acetate with sodium sulfate is unnecessary if the product is recrystallised from water anyways.
[EDIT 18.03.2022] From a quick test, ethylvanillin seems to have a decent solubility in toluene, so if the solubility data for protocatechuic aldehyde
in the section below are true this could be a very good solvent for recrystallisation. In water, ethylvanillin is less soluble than protocatechuic
aldehyde. In toluene, the reverse might be true - but I haven't found concrete data to confirm this.
Further notes:
Better TLC separation is obtained by toluene/ethyl acetate/acetic acid v/v/v 8/2/0.2:
Rf (ethylvanillin) = 0.52
Rf (protocatechuic aldehyde) = 0.19
However, the separation using petrol ether works well enough, and petrol ether is cheaper. For spotting the reaction mixture the addition of acetic
acid is not required as the solution is already acidic. For the final product AcOH should be included to reduce tailing. Staining with iodine vapor is
more sensitive than 254 nm UV. The TLC plates used were 5x10 cm silica gel 60 F254 glass-backed from Merck.
Ethylvanillin has an intense smell, considerably stronger than vanillin. The product doesn't have the same characteristic odor anymore, but it isn't
completely odorless either.
Instead of ethyl acetate, it is likely possible to extract with diethyl ether instead, as the patent does.
The solubility of protocatechuic aldehyde varies from source to source, but I found the following:
~ 50 g/L (cold water)[1]
~ 330 g/L (boiling water)[1]
5.0 g/L (0 °C) (water)[2]
11.7 g/L (15 °C) (water)[2]
1 g/mL in boiling alcohol[1]
very soluble in ether[1]
poorly soluble in boiling toluene[1]
nearly insoluble in cold toluene[1]
soluble in chloroform[3]
[1] - Beilsteins Handbuch der organischen Chemie, 4th edition, VIII, 1925, p. 246, full text on archive.org
[2] - J. Chem. Eng. Data 2012, 57, 7, 2018–2022, https://doi.org/10.1021/je300323g
[3] - Neues Handwörterbuch der Chemie, Band 5, 1890, p. 862, https://books.google.de/books?id=CqMEAAAAYAAJ
[4] - Purification of laboratory chemicals, 6th edition, 2009, p. 334
[5] - Verfahren zur Herstellung von Isovanillin, DE69507676
The main source/inspiration was German patent DE69507676:
Attachment: DE000069507676T2_all_pages.pdf (737kB)
This file has been downloaded 333 times
[Edited on 18-3-2022 by Diachrynic]
we apologize for the inconvenience
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi Diachrynic, very nice, well done!!!
So ethylvanilin is more reactive than vanilin. And also easier to obtain from shops (for ice cream production, for baking etc).
|
|
|
Diachrynic
Hazard to Others
Posts: 219
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Thanks Fery!
For me, both are fairly
easy to get, with ethylvanillin being slightly more expensive than vanillin. But nothing extreme. And the smell is worth it.
I did some more testing regarding the iron(III) complex. It is described in [1] as "green, and with addition of soda first purple, then red."
A spatula tip of protocatechuic aldehyde (left) and one of ethylvanillin (right) were placed in test tubes along with about 2 mL of distilled water.
An ultrasonic cleaner was used to speed up the dissolution. Two clear solutions are obtained:

Then, a drop of dilute iron(III) chloride solution was added. There is an immediate color change to dark green in the protocatechuic aldehyde
solution. The ethylvanillin takes on a very slight blue color. I diluted some of the green solution to show the color better.
 
Adding a solution of sodium carbonate to the small test tube causes a color shift towards orange-red:

Addition of dilute sulfuric acid to the green solution causes the color to fade and only leaves the faintly yellow color characteristic of Fe(III)
behind.
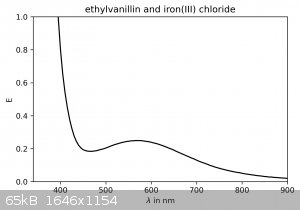
I couldn't resist taking some spectra of the dilute solutions (no concentration was measured) in the visible spectrum:
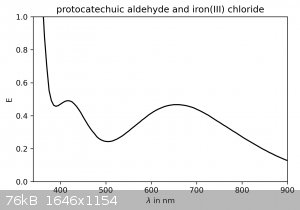 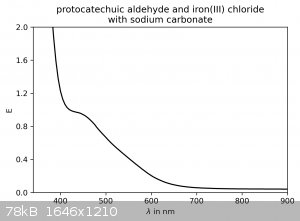 
For protocatechuic aldehyde with iron(III) chloride, peaks are observed around 415 and 655 nm. With the addition of sodium carbonate, a plateau around
425 nm is observed (it's not a true peak, probably due to the very high absorbance below 400 nm).
Ethylvanillin and iron(III) chloride show a peak around 570 nm.
These results, while giving further confirmation of the identity of the product, also reveal that on the TLC plates the protocatechuic aldehyde spot
may be visualized with a FeCl3-solution as a spray-stain. It will appear as a dark green spot. Ethylvanillin is not stained.
we apologize for the inconvenience
|
|
|
Texium
Administrator
Posts: 4508
Registered: 11-1-2014
Location: Salt Lake City
Member Is Offline
Mood: PhD candidate!
|
|
Nice work! I worked with 3,4-dihydroxybenzaldehyde a lot in my undergrad lab, as it was the starting material for my total synthesis project. I know
that sandy, light brown crystalline stuff well. That's what the commercially available stuff looks like- even from Sigma. I would be careful about
recrystallizing it. I tried a few times before and everything I did would just make it worse. The stuff really likes to oxidize. I quickly decided it
wasn't worth the effort, because the crude stuff worked fine in my reaction and the product I made with it (the di-TBS ether) was more stable and
easier to purify.
|
|
|
Pumukli
National Hazard
Posts: 686
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Very nice work, Diachrynic! I like these "simple", one step organic conversions. Especially if good smelling compounds are involved. 
I was surprised by Texium's comment. I read in an old article that those aromatic aldehydes that lack OH substituent on the ring are sensitive to
oxidation. For example benzaldehyde itself, but veratraldehyde (3,4-dimethoxy-benzaldehyde) too. On the other hand those that have free OH are much
more resistant. Vanillin (or ethyl-vanillin) can be kept "forever" in open air with very small change. Here comes Texium's comment on protocathecuic
being sensitive too...
Strange. Organic chemistry never ceases to amaze me.
|
|
|
Texium
Administrator
Posts: 4508
Registered: 11-1-2014
Location: Salt Lake City
Member Is Offline
Mood: PhD candidate!
|
|
It’s actually not the aldehyde that’s the problem in this case. It’s the catechol.
|
|
|
Pumukli
National Hazard
Posts: 686
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Yeah, resorcin also turns pink in itself by oxidation from air. No aldehyde group involved. Thanks for pointing that out.
|
|
|
Fery
National Hazard
Posts: 990
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Just an idea, but couldn't it be purified by vacuum sublimation in high vacuum so any oxidation is prevented?
|
|
|
Diachrynic
Hazard to Others
Posts: 219
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Quote: Originally posted by Fery  | | Just an idea, but couldn't it be purified by vacuum sublimation in high vacuum so any oxidation is prevented? |
According to J. Am. Chem. Soc. 1930, 52, 7, 2988–2994, https://doi.org/10.1021/ja01370a064 protocatechuic aldehyde can indeed be sublimated in high vacuum, providing "long white needles". They don't
provide a pressure but state that "a high efficiency mercury-vapor-pump" was used.
The same paper also gives some insight into how much toluene should be used for the recrystallization, 500 mL were employed for a bit more than 3.8 g.
On the note of solubility, Eur. J. Org. Chem., 2014: 111-121, https://doi.org/10.1002/ejoc.201301429 mentions that it is "barely soluble in dry DCM".
we apologize for the inconvenience
|
|
|
Texium
Administrator
Posts: 4508
Registered: 11-1-2014
Location: Salt Lake City
Member Is Offline
Mood: PhD candidate!
|
|
That’s just
not true. I ran my silyl protections in DCM and while it was rather slow to dissolve, it fully dissolved at a concentration of 0.25 M, and could
probably have been more concentrated than that.
|
|
|
Mateo_swe
National Hazard
Posts: 501
Registered: 24-8-2019
Location: Within EU
Member Is Offline
|
|
I quickly looked at that paper, it doesnt talk about protocatechuic aldehyde (3,4-dihydroxybenzaldehyde) or dihydroxy variants.
I does have a lot of info on 3,4-Dimethoxy-stuff though (and methylenedioxy).
Maybe the DCM solubility was for the methoxy.
I didnt read it very carefully though.
|
|
|
Diachrynic
Hazard to Others
Posts: 219
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
| Quote: Originally posted by Texium | | I ran my silyl protections in DCM and while it was rather slow to dissolve, it fully dissolved at a concentration of 0.25 M, and could probably have
been more concentrated than that. |
Okay, that sounds like it has a fairly high solubility in DCM then. Thanks
for the correction!
| Quote: Originally posted by Mateo_swe | | I quickly looked at that paper, it doesnt talk about protocatechuic aldehyde (3,4-dihydroxybenzaldehyde) or dihydroxy variants.
|
It's in the footnotes, number 23 states:
| Quote: | | Other aldehydes with additional functional groups were also studied: For example, 3,4-dihydroxybenzaldehyde and derivatives thereof, but they were
found to be barely soluble in dry CH2Cl2. |
Maybe "they" is referring here to the derivatives, but not the parent aldehyde? If so, then I (and Reaxys) was misinterpreting the text.
we apologize for the inconvenience
|
|
|
Diachrynic
Hazard to Others
Posts: 219
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Apparently this reaction is, in fact, already known in the literature, see JPH10265428A. I didn't know this when I tried the reaction.
Attachment: JPH10265428A.pdf (180kB)
This file has been downloaded 288 times
we apologize for the inconvenience
|
|
|
Mateo_swe
National Hazard
Posts: 501
Registered: 24-8-2019
Location: Within EU
Member Is Offline
|
|
Have you tried Aluminum Triiodide in DMSO?
The AlI3 is made in situ from aluminium powder and iodine.
Seems pretty good, only drawback is that much iodine is used and its a bit expensive.
Gram-Scale Synthesis of Protocatechualdehyde (7a)
To a mixture of AlI3 (38.5 mmol, 1.1 eq) prepared in situ from Al powder
(2.205 g) and I2 (14.655 g, 57.74 mmol) in MeCN (250 mL) was
added DMSO (3.009 g, 38.51 mmol, 1.1 equiv). The mixture was
stirred for 0.5 h at 80 °C, before vanillin (6a; 5.326 g, 35 mmol) was
added in portions. After stirring for 18 h at 80 °C, the reaction mixture
was quenched with aq 2 M HCl (50 mL), and extracted with EtOAc
(3 × 100 mL). The organic phases were combined, washed with sat. aq
Na2S2O3 and brine, and dried (anhyd MgSO4). After filtration, the organic
solvents were removed on a rotary evaporator, and the residue
was purified through flash column chromatography (eluent PE/EtOAc
11:9) to give 7a; yield: 4.413 g (91%).
Hmm, rename to xxx.pdf
It change finename of the attachment somehow.
Attachment: phpx5vw5T (463kB)
This file has been downloaded 309 times
[Edited on 2022-5-24 by Mateo_swe]
|
|
|
alexrandomkat
Harmless
Posts: 7
Registered: 23-1-2023
Member Is Offline
|
|
Gave this a try, been playing with doing this under an atmosphere without oxygen:
In a 25mL roundbottom, dry ice grains and 2.00g ethylvanillin powder were briefly stirred. 8.6 mL cold distilled conc sulfuric acid was added after a
waiting period, more dry ice grains were added in, and the flask connected to a trap and chain of five oil bubblers to maintain positive pressure in
the system (an earlier trial had suckback issues). Despite not being flushed enough by my judgement based on the volume of bubbles coming through, the
reaction stayed a translucent yellow for over half an hour under heating, before transitioning to dark green by an hour. The mix was heated in an 80C
oil bath for 4 hours, then poured over 16.5g ice with 8g DI water to give a dark green solution.
This aqueous solution was accidentally heated on the hotplate and allowed to get warm while stirring. The green solution turned reddish-purple during
that time. An additional 13.4g ice was added. The liquid was extracted with ~5-8mL ether portions until ~40mL of clear reddish extract was obtained
(same color as Diachrynic's image). The extract was dried over magnesium sulfate, then, the following day, the ether boiled off under atmospheric
pressure until a small amount of liquid remained, and finally stirred in a 65C oil bath under vacuum (water aspirator), which led to the crashing out
of a light-pink slurry. Further stirring and heating under vacuum gave a fine, free-flowing off-white powder. 1.16g of product (after a small amount
was used for TLC) remained, a yield of 70%.
Something to note, when extracting with diethyl ether much more of the tar seems to come out. An attempt with 8g ethylvanillin where I failed to
exclude oxygen (attempted cycling vacuum with butane purges from a balloon, clearly my technique was abysmal. Also a little water sucked back from my
water aspirator into the whole thing.) gave a reaction mix that looked like Diachrynic's image (reaction instantly turned black upon stirring in the
acid under heating). After pouring over ice and extraction with ether, the extract was an opaque black liquid. Concentration (incl. heating under
vacuum) left me with a black oil which crystallized out into a brown substance upon standing. In this attempt some water drops also got into the
reaction from suckback from the water aspirator... Heating was similar, 75-80C for 5hr.
Reaction setup using dry ice:

Two results (0 is the crystallized black oil from 8g ethylvanillin, 1 is the result of the CO2 atmosphere from 2g ethylvanillin):

sad TLC under 254nm bulb (2.3 : 2.3 : 0.2 - EtOAc : hexanes : AcOH, E is ethylvanillin, 0 and 1 are same as above)

(I had one using a ratio 7:2:0.2 that had worse separation, and also just looked abysmal bc I spotted waaaaay too much. Got a mushroom cloud on the
TLC. Maybe would make a boring art piece.)
I'm thinking most of my lost yield was from spilling a bunch of the aqueous solution on the ground during extraction lol. Given I only put small
amounts of dry ice in that I scraped off a block with a spatula, it seems plausible much better results could be obtained if one added an actual chunk
in and used enough to properly flush the flask. TL;DR -- be better than me.
------
Questions:
Not that this reaction needs to be changed for any practical purposes, but I'm wondering if this can be sped up with the addition of a very small
amount of NaBr? My understanding (based on guesswork) is that we're doing an Sn2 with the HSO4- attacking the protonated ether. I assume that's the
reason why the original source brought by Diachrynic uses a round 1:2 molar ratio of ethylvanillin:water if you assume 98% sulfuric acid, and not any
less.
I assume the reaction doesn't tolerate water well (as seen by the big ethylvanillin splot on the TLC of 0, assuming that's because of the water from
the aspirator coming in at the start ruining things). Am curious why, as at first glance it seems that the cause would be the formed hydrogen ethyl
sulfate hydrolyzing back to ethanol which would undergo dehydration with the product. But that makes me question why using a large excess of conc
sulfuric as I did with my successful trial still gives good results, since with that extra sulfuric brings extra water. All that extra water is still
fully protonated though, so does that mean there exists a lower bound of tolerable concentration, below which the reaction dies? I don't have much of
a good idea how much water sucked back into my reaction. Or maybe the reaction does tolerate water well and that trial just failed for other reasons.
|
|
|
|









