SyntheticFunk
Harmless

Posts: 21
Registered: 30-12-2022
Member Is Offline
|
|
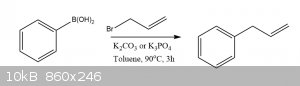
Synthesis of (Prop-2-en-1-yl)benzene: A Catalyst Free Suzuki-Like Coupling
Pardon the IUPAC name, the intent is to make it slightly more difficult for cooks to come across. Not that I care how you spend your time or what you
do with this, individuals should be able to do as they wish within the privacy of their own home so long as it does not harm others. It is just that I
do not like it when those who do not really care about the art try and find a quick and easy way to exploit it for personal gain at the expense of the
community as a whole. But I digress.
Alternative Methods of Production
The three methods I most often encounter for the synthesis of propenylbenzene is either:
1. Grignard reaction between a bromo- or iodobenzene and allyl bromide.
2. Friedel-Krafts Alkylation of Allyl Bromide
and less frequently
3. Friedel-Krafts Alkylation of Allyl Alcohol
The Grignard reaction suffers from relative operational complexity (inert atmosphere, dry solvents, etc) while resulting in only modest, though
ultimately acceptable yields.
The Friedel-Krafts Alkylation is an appealing alternative, however, it too has a few drawbacks. The most obvious being the use of carcinogenic
benzene. Another drawback in addition to this hazard is the further reaction of the olefin product with HBr to yield either the hydrobrominated
product (2-bromo-1-phenylpropane), or polymerized crap from reaction with other olefins. In my experience, the use of ZnCl2 as a catalyst
reduces the extent of hydrobromination compared to when the stronger relatives AlCl3 or FeCl3 are used.
Lastly, the use of allyl alcohol may appear enticing at first due to the ease of generating the allylic carbocation in acidic conditions, however,
this will almost certainly lead to extensive side reactions of polymerization. Perhaps this can be minimized with some optimization, but with lower
hanging fruit available, I would argue there is little point. I seem to recall success with the reaction of benzene and allyl alcohol using ZnCl2, but
I cannot find my notes on it and therefore cannot be certain nor provide conditions. I've included an article that performs this synthesis at the
bottom of this post.
Catalyst Free Suzuki-Like Coupling
There are several factors that contribute to this method's superiority over the aforementioned.
1. Short reaction time (3hrs)
2. Use of commonly available solvent (toluene)
3. Moderate Temperature (90C)
4. No inert atmosphere needed/Operationally simple
5. Relatively good yields (80% reported in literature, 71% obtained herein on a 20 gram scale).
6. Commercial availability and stability of boronic acid salts.
7. Very few byproducts formed and consequently a simple(ish) purification
Since using a Grignard is one of the most common ways to prepare organic boronic acids, one could fairly criticize this method as an unnecessary
additional step. However I would counter that there are alternate ways to prepare arylboronic acids that do not require a Grignard reaction and the
stability of the arylboronic acid allows for long term storage with ease. Furthermore phenylboronic acid is commercially available.
Drawbacks:
1. Large solvent volume
2. High boiling point of toluene slows down removal
3. Significant foaming during solvent removal
Though the amount of solvent used can likely be reduced with some effort towards optimization, I used a similar ratio of reactants : solvent as the
authors (2 mmol reactant : 10mL of toluene), but rounded down to the nearest half litre to save space and time spent removing solvent at the end.
Substituting potassium carbonate for potassium phosphate is tolerated, though the authors note other bases result in lower yields.
Without further adieu, let's get into it.
To a 2L round bottom flask equipped with a reflux condenser and a stir bar was added 1.5L of toluene, 20g of allyl bromide, 20g of phenylboronic acid,
and 34g of K3PO4. The reaction was heated to 90oC and stirred for 3 hours before being allowed to cool to room
temperature.
An attempt was made to distill the mixture with a small fractioning column to remove the toluene, however a significant amount of foaming hindered
this process. The mixture was poured through a small amount of celite then dried with sodium sulfate to reduce foaming and was subsequently evaporated
to near-dryness on a rotary evaporator giving slightly yellow mass comprised mostly of boric acid and potassium phosphate salts. The mass is extracted
4x with 50mL of 10% ethyl acetate in hexanes, the extracts pooled, and concentrated under reduced pressure to yield a yellow oil weighing 13.7g (71%
of theory). A small (~1mg) quantity of product was analyzed via gas chromatography-mass spectrometry which indicates very few byproducts are present.
Thus, no further purification was attempted. The authors, however, purified the product further chromatographically on silica gel (diethyl ether :
n-hexane 1:9).
NOTE: The authors recrystallized the phenylboronic acid beforehand to hydrolyze any phenylboronic anhydride into the corresponding acid as they noted
that the reactivity varied depending on the quantity of anhydride. I did not bother with this and added a small quantity of water (undetermined, just
added through a squirt bottle) as an alternative. This water is removed azeotropically during evaporation or by sodium sulfate/celite.
Apologies for the lack of details on certain components. This was performed a few months ago and my notes lack various details. Overall, the reaction
was incredibly simple to perform and the product was used successfully for a subsequent reaction without any purification required.
GC-MS of product
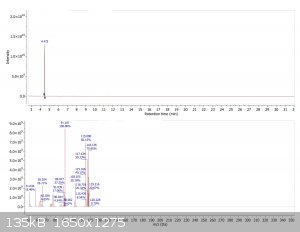
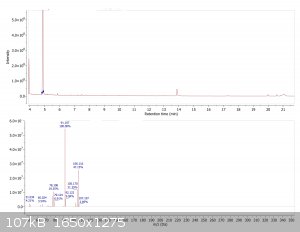
A closer look at the impurities. Typical impurities expected of a Suzuki coupling include homo coupled starting material, as well as deboronated
starting material. The m/z values of the impurity peaks correspond to such products. NIST search also revealed either dimethylbenzene or ethylbenzene
as a minor byproduct (mass spectrum shown below corresponds to the earliest eluting peak only)
Attachment: propenylbenzenesuzukilike.pdf (326kB)
This file has been downloaded 118 times
Attachment: allylbenzeneallylOH.pdf (193kB)
This file has been downloaded 117 times
[Edited on 16-5-2023 by SyntheticFunk]
[Edited on 16-5-2023 by SyntheticFunk]
|
|
|
clearly_not_atara
International Hazard
Posts: 2692
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Arylboronic acids can also be obtained as the esters by electrosynthesis with trimethyl borate:
https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
[Edited on 04-20-1969 by clearly_not_atara]
|
|
|
SyntheticFunk
Harmless
Posts: 21
Registered: 30-12-2022
Member Is Offline
|
|
Thanks for contributing this! Wasn't aware. I've read that the preparation of organic boronic acids aren't too difficult per se, it's the work up and
isolation. Im not sure how valid that statement is, but I would imagine an electrochemical based synthesis would, at least partially, alleviate this
problem as it employs less reagents to separate in the end.
Would love to hear anybody else's thoughts on this matter or matters related.
Thanks again for the contribution.
|
|
|
clearly_not_atara
International Hazard
Posts: 2692
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
The paper you uploaded mentions that the arylboronic acids are best used "wet", by suction drying from water, since they will oligomerize when
isolated. I assume that this would alleviate the difficulties. The dimethyl boronate esters produced by the electrosynthesis are likely to rapidly
hydrolyze in aqueous solution giving the free acids, due to the tendency of boron to form four-coordinate anionic species. The attached paper
discusses the hydrolysis of boronate esters, but mostly in the context of attempting to develop boronates that resist hydrolysis.
However, the paper you linked also shows slow reaction rates and poor yields (Table 2) with the unsubstituted allyl bromide. It appears (Table 1) that
using an excess of the electrophile gives a better yield, which is fine here, since the allyl bromide is obviously the cheaper half of the reaction.
Allyl chlorides are said to react slower, and while it wasn't tested, simple extrapolation suggests that allyl iodide may be a better substrate.
The usual non-electrosynthetic preparations of boronates usually involve organometallic precursors or boron halides. I assume that this contributes to
the difficulty in work-up, since these can be rather poorly behaved.
Overall, nice find.
Attachment: bernardini2009.pdf (71kB)
This file has been downloaded 100 times
[Edited on 04-20-1969 by clearly_not_atara]
|
|
|
|








