| Pages:
1
..
6
7
8
9 |
andyloris
Harmless

Posts: 49
Registered: 22-9-2022
Location: France
Member Is Offline
Mood: Lazy
|
|
Then could this work with aspartic acid ?
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Since acrolein preparation is no joke and yields are not great in laboratory scale procedures as much tar is produced too, is making malonic acid via
acrolein (which I assume you are looking at because it can be prepared from easily available glycerine and sodium bisulphate) really an amateur
friendly route?
As andyloris points out there are several well explored routes from aspartic acid and also through beta-alanine. Both of which are cheap and degraded
through well studied wet chemistry.
Also what's wrong with the route through 1,3-propandiol as this compound is now easily available online through hobbyist cosmetic ingredient
suppliers, particularly in the US but they posted it to me in the UK without problem.
|
|
|
andyloris
Harmless
Posts: 49
Registered: 22-9-2022
Location: France
Member Is Offline
Mood: Lazy
|
|
What is the route from beta-alanine ?
|
|
|
Texium
Administrator
Posts: 4508
Registered: 11-1-2014
Location: Salt Lake City
Member Is Offline
Mood: PhD candidate!
|
|
Perhaps you could try using a search engine instead of asking so many one-lined questions in rapid succession.
|
|
|
andyloris
Harmless
Posts: 49
Registered: 22-9-2022
Location: France
Member Is Offline
Mood: Lazy
|
|
I searched on google but I only could find a source saying
| Quote: |
beta-Alanine can undergo a transamination reaction with pyruvate to form malonate-semialdehyde and L-alanine. The malonate semialdehyde can then be
converted into malonate
|
However I thought a transamination reaction was the swaping of a ketone of a keto acid and the amine of an alpha-amino acid to form a
new amino acid and a new keto acid. However beta-alanine is a beta amino acid, and this didn't make sense.
[Edited on 25-10-2022 by andyloris]
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi andyloris, give me a few days and I will track down the various references but I seem to recall that hypochlorite ions or their proxies such as the
chlorimines like TCCA and NaDCCA convert b-alanine to hemialdehydo-malonate ion. I have several papers at home on these lines.
|
|
|
SplendidAcylation
Hazard to Others
Posts: 196
Registered: 28-10-2018
Location: Starving in some deep mystery
Member Is Offline
Mood: No one I think is in my tree.
|
|
Quote: Originally posted by andyloris  | Just got a random idea while looking at the strecker degradation wikipedia page:
Could serine undergo strecker degradation to form 3-hydroxypropanal and then be oxidised to malonic acid ? |
I looked at this and I thought the number of carbons was right too 
So easy to miscount.
| Quote: |
Since acrolein preparation is no joke and yields are not great in laboratory scale procedures as much tar is produced too, is making malonic acid via
acrolein (which I assume you are looking at because it can be prepared from easily available glycerine and sodium bisulphate) really an amateur
friendly route? As andyloris points out there are several well explored routes from aspartic acid and also through beta-alanine. Both of which are
cheap and degraded through well studied wet chemistry. Also what's wrong with the route through 1,3-propandiol as this compound is now easily
available online through hobbyist cosmetic ingredient suppliers, particularly in the US but they posted it to me in the UK without problem.
|
Yes, my primary motivation was based on the easy synthesis of acrolein from glycerol.
That's interesting, I wasn't aware of these routes, are they theoretical or are they suspected to work?
Indeed, there isn't anything particularly pleasant about acrolein, but, having read all of the threads I could find here regarding malonic acid, I
couldn't see any practical routes avoiding cyanide; Even the oxidation of malic acid seems to be fruitless.
I would be much happier handling cyanides than acrolein, that's for sure, but cyanides are not easy to come by, and if I were to acquire some, I doubt
I'd waste them on the synthesis of malonic acid, as you say, 1,3-propanediol is probably the most feasible route.
There has been much talk about the synthesis of cyanides using chloroform and ammonia, but very few positive results, so it seems quite unlikely to be
successful.
I shall await your references! 
|
|
|
andyloris
Harmless
Posts: 49
Registered: 22-9-2022
Location: France
Member Is Offline
Mood: Lazy
|
|
The route from aspartic acid is a random idea I got, but I see no reason why it wouldn't work.
|
|
|
kmno4
International Hazard
Posts: 1495
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
..... that is why some time ago I decided to order malonic acid directly from a manufacturer (PRC). Unfortunately, shipping costs almost doubled total
price, giving ~40 $ per 1 kg of malonic acid in the end.
Earlier I made this acid by standard method (chloroacetic + cyanide), but without ether continuous extraction apparatus, preparing the free acid is
highly problematic. Besides, using K/Na cyanide for this purpose is a waste. There are many better ways to utilize the cyanide 
Currently, malonic acid is cheaply (let us say ...) prepared in industry by fermentation methods.
Слава Україні !
Героям слава !
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
While I agree that malonic acid is one of those simple organic building blocks that is best bought I can't help but feel that the challenge has a
certain attraction for masochists .
So a few days ago I decided to try a route from beta alanine. First I tried just dissolving 10g of beta alanine in a molar equivalent fairly strong
sodium nitrite solution (7.75g in 50ml of water) and then gently warmed it. When the temperature reached about 60-70 C a slow evolution of nitrogen
began but it was very slow so I added a molar equivalence of 50% nitric acid fairly quickly. The rate was chosen so that the gas remained colourless
(hopefully therefore nitrogen), too rapid an addition caused the gas to become brown. The temperature rose to about 90 C and the evolution of gas was
vigorous. When the reaction subsided I evaporated the solution down to about 20-25ml on a steam bath and let it cool. About 6g of sodium nitrate
rhombs crystallised out, they were filtered off but weighed damp. The filtrate was warmed to about 70 C and the 40ml of 50% nitric acid added
dropwise. It took quite a while before any reaction started but when it did the mixture boiled steadily throughout the addition, much brown NO2
evolved. When the reaction subsided it was cooled in an open bowl to allow further evaporation. The pale orange yellow liquid was chilled overnight
but no crystals formed so it was neutralised with 50% NaOH solution, still no crystals formed so the solution was evaporated from about 40ml to 20ml;
copious colourless crystals appeared as evaporation occurred. When no further evaporation appeared to occur the slurry was cooled and then filtered,
The cake comprising almost colourless crystals weighed 6.47g is yet to be investigated and a viscous orange-brown solution which is currently being
evaporated further on a hotplate.
The reaction of nitrous acid with beta-alanine yield 3-hydroxypropanoic acid and nitrogen but without a mineral acid it is very slow. The former
dehydrates easily to acrylic acid but I am not sure how easily this occurs in aqueous solution or how important this is given that the intermediate
product wasn't isolated but th reaction temperature at times reached boiling point. However, the viscosity of the final filtrate suggest that some
polymerised acrylate like material is present. My work-up plan is to treat the latter crystals with a little nitric acid, evaporate to dryness on the
steam bath and leach out any malonic acid with alcohol. Watch this space.
I may try this reaction again using beta alanine and 50% nitric acid with just a catalytic amount of sodium nitrite to generate the initial nitrous
acid. I am also going to try Magpie's 1,3-propanediol method now I have the diol.
[Edited on 28-10-2022 by Boffis]
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Well the proposed nitric acid oxidation of beta alanine with a trace of nitrite to act as catalyst seems to work nicely, although I haven't
characterised the product yet. I tried various methods but the best seems to be to moisten 10g of beta-alanine with a few ml of water and warm it on a
steam bath in a ceramic bowl. When it is hot to the touch, c 70 C, remove from the heat and add 11-12ml of conc. nitric acid slowly, drop by drop at
such a rate that the frothing is manageable and only the slightest brown NO2 is generated and the temperature runs at around 90 C. Have a bowl of cold
water available to cool the reaction if it gets too frisky. When the addition of nitric acid no longer provokes vigorous effervescence the reaction is
done. Return to the bowl to the steam bath and steam for a couple of hour to drive of most of the remaining water to leave a viscous yellow liquid
which solidifies completely on cooling.
I am currently researching the recrystallisation of malonic acid; has anyone got any experience of recrystallisation solvents for this acid. I have
checked out "Purification of laboratory chemicals" but they recommend water, acetone or a complex mixture of benzene etc.? Water would be nice but the
solubility is so high a lot will be left in the filtrate. Anyone got experience of acetone?
|
|
|
kmno4
International Hazard
Posts: 1495
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Boffis |
I am currently researching the recrystallisation of malonic acid; has anyone got any experience of recrystallisation solvents for this acid.
|
I do not have such experience, but there are many papers giving solubility of malonic acid in many solvents and their binary mixtures (THF, IPA,
ethanol, EtAc, dioxan, MeCN, acetone... etc)
For example here:
https://doi.org/10.1002/ceat.201700227
Слава Україні !
Героям слава !
|
|
|
Pumukli
National Hazard
Posts: 686
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
I could not resist and tried to make malonic acid via the nitric acid oxidation of malic acid.
Well, it was a failure as far as I can tell.
No oxidation took place, at least nothing obvious (no NOx development), though I tried to "boost" the process with a few milligrams of NaNO2 too.
The acid was not concentrated, only around 50-55%, but the temperature was above 70 Celsius.
|
|
|
kmno4
International Hazard
Posts: 1495
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
I made some literature research, no procedure I found describes formation of uniform product in case of oxidation of aminoacids.
Becuse strong acid is used, the first step is formation of ammonium salt, formation of some diazo compound in this case is highly doubtful and if
even, it does not explain further reactions.
Besides, oxidation of some organic compounds by HNO3 gives N2O as product of nitrogen reduction - it may be mistaken with N2.
As I said earlier, formation of complex mixture of products, even if more malonic acid is formed, makes further work up cumbersome.
And the more HNO3 is used in higher temperatures, the more (COOH)2 is always formed.
Слава Україні !
Героям слава !
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@ kmno4; Well I have something to report but I am not quite sure what!!
I ran a 20g batch of beta-alanine and obtained a workable amount of beautiful colourless crystals up to 30mm long. It melts without decomposition if
heated carefully to a clear liquid and then as the temperature climb gives of gas an decomposes. I heated quickly on foil it melts then suddenly puffs
away. I have set up a melting point jig so I'll run a more accurate test tomorrow but the Mp appears to be <90 C. (beta alanine 207, Malonic acid
135)
My original idea was that the presence of nitrous acid or a proxy would replace the amine group with a hydroxy group producing 3-hydroxypropanoic acid
would could them be oxidized by the nitric acid. The production of reduced nitrogen oxides as the oxidation proceeds would convert more beta alanine
to the hydroxy acid and so the reaction should be self-sustaining until the amino acid is used up. However, the product does not appear to be malonic
acid.
In my searches I came across a couple of interesting examples of nitric acid oxidation of simple esters (eg ethyl acetoacetate) to produce furazan or
furazan-2-N-oxide derivatives. These compounds often have fairly low Mps and are often energetic.
Other possibilities are things like tartronic acid (hydroxymalonic acid Mp 157) but the Mp is too low.
By the way if you try these experiments don't try recovering the wash acetone from the recrystallisation. When the acetone is distilled it results in
a violent decomposition rather suddenly in spite of the fact that free nitric acid has been almost totally removed. The impurity in this reaction is a
water soluble yellow oil that deflagrates quite readily!!
The plot thickens!
|
|
|
kmno4
International Hazard
Posts: 1495
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
Oxidation of organic matters by HNO3 has free-radical mechanism and is not very selective. Of course, same HNO3 is not responsible for hydrogen
abstraction from the organic molecule (as it happens usually), but its oxides do this (because NO or NO2 acts as radicals, but N2O does not).
Additional complications is nitration and nitro-esters formation when too concentrated HNO3 is used. Usually, concentration below 20% is used, to
avoid such reactions. Very common is 10% HNO3 as oxidation agent.
Of course, there are many organic compounds giving large yields of desired oxidation products even when conc. HNO3 is used.
But the dependency of oxidation-nitration properities of HNO3 (depending of its concentration) is well established.
As I wrote above, -NH2 group is protonated under acidic conditions and becomes partly inert. For example, reaction of phenylalanine with aq. HNO3
gives p-nitrophenylalanine in rather good yield, -NH2 is retained.
Слава Україні !
Героям слава !
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Well, carried out a Mp test on my white crystals and got a figure of 93-93.5 C; the compound melted sharply and without decomposition. Any ideas? The
sharp melting point suggests a pure compound and it is obtained in high yield; 20g of beta-alanine gave 31g of product and after recrystallisation and
washing in acetone 26g, this is far too high for any simple oxidation product it must incorporate some part of the nitric acid. Clearly this is a very
complex reaction but worthy
By the way if you think furazan derivatives (Oxodiazoles) or furoxans (Oxodiazole-N-oxides) are not that likely check out some of the threads on SM
and in particular a reference posted by Dany:
1] Snyder, H. R., & Boyer, N. E. (1955). The Synthesis of Furoxans from Aryl Methyl Ketones and Nitric Acid1. Journal of the American Chemical
Society, 77(16), 4233-4238. doi: 10.1021/ja01621a021
|
|
|
Texium
|
Thread Topped
9-11-2022 at 13:20 |
Texium
|
Threads Merged
9-11-2022 at 13:21 |
kmno4
International Hazard
Posts: 1495
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Boffis | Well, carried out a Mp test on my white crystals and got a figure of 93-93.5 C; the compound melted sharply and without decomposition. Any ideas?
|
I can be everything (almost).
But if you like furoxan-like matters, here you are a candidate:
furoxancarboxylic acid (I am not sure if it is correct name)
https://archive.org/download/crossref-pre-1923-scholarly-wor...
.... and find page 66. The structural formula is not fully correct in this old paper: the oxygen bridge is not good.
Слава Україні !
Героям слава !
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi kmno4, thankyou for the paper, I haven't seen this one before but I have looked at, and indeed translated several others on "glyoxime peroxides" as
part of my investigation into fulminic and fulminuric acid.
I have found online a book on the furazan and furoxan heterocyclics and there are lots of examples of nitric acid oxidation of various ketones and
keto-acids. I have attached a copy of the Synder paper.
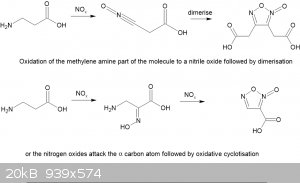
Looking at the published literature there are two possible reaction routes but neither is quite applicable to a beta-aminoacid. In one route the
nitrogen oxides attack the amino group and somehow generate a nitrile oxide rather than a diazo compound. I can't find any info on the
furoxan-diacetic acid. My original idea was that a transient diazo compound formed that then hydrolysed to the hydroxy acid which was then simply
oxidized to a second acid group is not tenable in view of the results.
In the second, more likely, route nitrous acid from the nitrite attacks the alpha carbon to the carboxylic acid group forming a isonitroso group while
the amine is oxidised to a nitroso group that rearranges it the isonitroso form and is then oxidised and cyclotises to the furoxan carboxylic acid. I
prefer this latter route.
Attachment: The preparation of furoxans by oxidation of methylketones with nitric acid JACS 1955.pdf (640kB)
This file has been downloaded 212 times
[Edited on 11-11-2022 by Boffis]
|
|
|
kmno4
International Hazard
Posts: 1495
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
The "diaceto" derivative would have higher m.p. than the monocarboxylate furoxan, at least I think so.
You may do some NaOH titration of your compound and/or make some tests described in this article from Annalen. The most possibly your preparate is
purer and gives higher m.p. than noted there (89-91 C). The Ag salt is "explosive", as noted elsewhere, but many Ag salts of "nitrogen-acid" do so.
I would name your reaction as "Boffis reaction", but somehow I have impression that it is too simple and someone must have done it earlier. However, I
think that making something new, a reaction or a compound, is not very difficult even at "home-chemistry" level.
The chemistry is the endless sea of possibilities
P.S. I am going to do some additional literature research, but maybe SciFinder or similar tool would be more helpful (I have no access now)
[Edited on 12-11-2022 by kmno4]
Слава Україні !
Героям слава !
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I agree with you kmno4 about the mp of the diacetic acid and I find this route rather implausible too. Apart from Synder's paper I have read many
papers that produce what are clearly furoxans, although usually referred to as glyoxime peroxides, with nitric acid including the original paper by
Propper and several by Ulpiani. Unfortunately other interesting paper by Ponzio and Quilico are in issues of Gaz. Chim. Ital that are not readily
available (up to 1923 is available at archive.org but these two latter authors mostly published around 1926 to 1938. I don't have access to scifinder
either, any volunteers out there that do?
I have just been preparing salts of the compound and got some weird results that suggest the compound is not furoxan carboxylic acid. It does not
appear to be decomposed by the amount of NaOH required to neutralise it and the resulting solution can be evaporated on a steam bath. There is no ppt
with either ammoniacal silver nitrate or 2M silver nitrate solution with either the free acid or the neutralised acid and the barium salt is also very
soluble.
I originally thought that it might be a compound oxidised in the central carbon like tartronic acid or mesoxalic acid but the former melts at 157 and
the latter is so unstable that it can't be isolated in solid form but could it be a nitrate ester of either tartronic acid or 3-hydroxypropanoic acid?
I am also becoming interested in the yellow energetic, water soluble oil that accompanies it after reading that many of the furazan and furozan
compounds are viscous liquids. I am being sent overseas with my work soon so I will loose access to my lab until Christmas so I am running out of
experimental time.
|
|
|
kmno4
International Hazard
Posts: 1495
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
OK.
As a matter of fact, it is really hard to guess correct structure of the product without elemental analysis, IR spectra.... etc.
I would make a test with NaHCO3 sol. to see if your substance is acidic (carboxylic) at all. The crystal form may also suggest some oxime, many of
them form large long crystalline needles. Possibly a test with Ni salts (in alkaline conditions) would also show something. Besides, its Ag or Ba
salt can be water soluble, who knows....
BTW, the yellow oily matter can be also whatever: some nitro derivatie, nitrate ester, nitrosoamine ... etc, also in multisubstituted versions.
[Edited on 13-11-2022 by kmno4]
Слава Україні !
Героям слава !
|
|
|
Boffis
International Hazard
Posts: 1836
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi kmno4. I haven’t replied to this thread earlier as when at home over the Christmas period I was rather ill and didn’t really feel like doing
any chemistry and I am now overseas again for a while. However, I have been doing some reading into this subject and found some old but rather
interesting papers. You are quite right, I am just guessing about the identity of the my mysterious compound but not having access to physical
measurements other than melting point my method of working is; “guess the identity then check the literature for its properties, do they match;
-yes, whoopee –No, back to the guessing game” technique.
You might think that every possible C3 derivative of propanoic and malonic acids has been prepared but much of the information on these compounds is
very old and many have not been isolated in a satisfactory solid form. One such compound is the theoretical 2-oxime of mesoxalic acid semialdehyde.
Van Slyke developed a method1,2) for the determination of amino-acid amino groups by treating the acids with sodium nitrite in acid solution. Some
amino acids give slightly anomalous results and during a follow-up investigation by Schmidt4) it was found that beta-alanine reacted like a normal
amino acid and rapidly de-aminated, presumably to beta-hydroxypropanoic acid though this isn’t expressly stated. The reaction is essentially
complete in 3-5 minutes. So if 1,3-propandiol is oxidized by nitric acid to malonic acid one would expect the hydroxylpropanoic acid to be an
intermediate and yield the same product, in fact this is expressly stated in von Richter’s textbook of carbon compounds. So I would expect malonic
acid to be the end product of nitric acid oxidation. However, in my final experiment I didn’t add any sodium nitrite so the oxidation route is much
less clear. One slight complication is that beta-hydroxypropanoic acid, which is a well-studied intermediate for bio-polymers(see Wikipedia), readily
dehydrates to acrylic acid and this may react differently towards nitric acid too. A further problem I see is that an oxime group in the 3 position
allows it to cyclotise to an oxazolone derivative.
When I am home next I will try nitrous acid de-amination of beta-alanine then nitric acid oxidation in separate stages to see if I get the same
material. I may also try repeating Magpie’s procedure to see what I get.
I also intend to try treating the product with hydroxylamine to see if it has any carbonyl groups in it.
While rummaging through the old literature I came across another paper investigating the reason for the excess nitrogen by Austin3) in which he
concludes that one of the products of de-amination of glycine with nitrous acid are things like methazonic acid and fulminic acid and that the
polymerization and decomposition of these material leads to the formation of excess nitrogen. Incidentally this paper has been posted before, I think
it was on chemplayer’s thread on "essence of smirf" which is described in the latter part of this paper.
1 The reaction of alpha amino acids and related with nitrous acid JBC Van Slyke 1911 v09 p185
2 Quantitative determination of aliphatic amino groups JBC Van Slyke 1912 v12 p275
3 Deamination of amino acids by nitrous acid JCS A T Austin 1950
4 The reaction of nitrous acid with certain amino acids at 45 C JBC C L Schmidt 1930 v82 p587
References 1,2 and 4 are available from the Journal of biological chemistry web site freely.
Attachment: 3 Deamination of amino acids by nitrous acid re methylnitrolic acid & fulminates JCS A T Austin 1950.pdf (205kB)
This file has been downloaded 193 times
|
|
|
Grizli7
Harmless
Posts: 39
Registered: 1-9-2021
Member Is Offline
|
|
Is malic acid + hydrogen peroxide interesting?
|
|
|
dicyanin
Hazard to Self
Posts: 57
Registered: 29-3-2020
Location: Europe
Member Is Offline
Mood: inquisitive
|
|
regarding the oxidation of malic acid w. ammoniacal H2O2
| Quote: Originally posted by Magpie | I have recently tried a preparation of calcium malonate using an older procedure by Subramaniam et al, as offered by DJF90. This procedure, as
excerpted, is shown below:
"Oxidation of Malic Acid by Hydrogen Peroxide in the Presence of Ammonia.
dl-Malic acid (5 g.) was dissolved in a 20-volume (6%) solution of hydrogen peroxide (100 c.c.), and the liquid was made just alkaline with
concentrated aqueous ammonia and diluted to 150 c.c. with water. After 30 hours the reaction mixture was shaken with animal charcoal (5 g.) to remove
the undecomposed hydrogen peroxide still present. The solution was filtered, boiled, and treated with an excess of calcium chloride solution. After
concentration to 100 c.c. and subsequent standing for 3 days, the white crystalline precipitate (5.25 g.) was collected, washed with water, and dried
in a vacuum [Found : Ca, 21.7. Calc. for CH2(CO*O)2Ca,2H2O: Ca, 22.3%].
A quantity of this calcium salt (1.5 g.) was heated in a sealed tube with glacial acetic acid (5 c.c.) and freshly distilled cinnamaldehyde (30 drops)
at 100° for 10 hours. The product was purified as detailed in a previous case and yielded 0.32 g., which melted at 208° (decomp.), alone or mixed
with authentic cinnamylidenemalonic acid of m. p. 208° (decomp.) (0.1510 g. required 27.4 c.c. of N/20 NaOH. Calc., 27.7 c.c.)."
A white precipitate of the stated weight was indeed obtained. However, extraction of the acidified salt with ether did not yield the desired
malonic acid.
Although a method for identifying the salt as a malonate is presented in the article, it appears cumbersome. Does anyone have any other suggestions
as to how putative Ca malonate can be identified? |
I have repeated the above procedure 4 times (although using L-malic acid from a brewery supply store), and in each occasion about 4-5 grams of white
precipitate was obtained after the addition of an aqueous CaCl2 solution. Although I had no benzenediazonium solution at hand to qualify
the formation of malonic acid as stated in the paper, and just let the reaction go for exactly 30 hours in every case. However, just like our late and
lamented member Magpie suspected, I have found the calcium salt precipitate contains a mixed product and the actual malonate content of the
precipitate is rather low.
The main problem is that the method in the Subramaniam et al. (1929) paper, although reproducable, is presented in a somewhat dishonest manner. The
white precipitate was assumed to be calcium malonate dihydrate, the author analysed the Ca content and found 21.7%, compared to the theoretical 22.3%.
Note that he did not analyse/report C, H and O content.
I repeated their derivatisation by mixing 1.5 grams of the dry precipitate with 5 ml glacial acetic acid in a glass tube, added 30 drops of
cinnamaldehyde, sealed the tube and heated it in an oil bath at 100°C for 10 hours.
Because they provided no details for the workup, after checking the literature a more recent paper, Hoogen & Nuyken (2000), was found where they
prepare cinnamylidenemalonic acid in a similar fashion, but by adding a catalytic amount of pyridine their reaction was finished after 90 minutes.
They reported 58% yield (much higher yields were obtained when methoxy-substituted cinnamaldehydes were used, so the modest yield is specific for this
substrate).
After heating the sealed tube for 10 hours, yellowish-white solids and a red supernatant liquid phase was observed. Following the Hoogen & Nuyken
(2000) workup procedure, the contents of the tube were shaken well, then added to a solution of 25 grams Na2CO3.10H2O
in 50 ml demineralised water. After foaming subsided, the excess cinnamaldehyde was removed by washing with 25 ml diethyl ether. The pH of the aqueous
phase was about 9-10 (indicator paper). It was gently acidified by the careful addition of azeotropic aqueous HCl until a pH of about 3 was reached.
There was observed a small amount of precipitate as yellowish needles. After standing overnight in near freezing temperatures, the solids were
collected by filtration, they were air dried then weighed: 0.10 g (there was more but part of it was absorbed by the filter paper).
Subramaniam et al. (1929) reported a yield of 0.35 g cinnamylidenemalonic acid from 1.5 g calcium salt, and a mp of 208°C. Hoogen & Nuyken (2000)
reported an mp of 191°C for their product, which they first washed with hot benzene to remove cinnamylideneacetic acid formed during the reaction in
small percentage (presumably due to thermal decarboxylation), and subsequently dried and recrystallised from ethanol.
malonic acid MW = 104.061 g/mol
calcium malonate MW = 142.119 g/mol
Ca malonate.2H2O MW = 178.149 g/mol
cinnamaldehyde MW = 132.16 g/mol
cinnamylidenemalonic acid MW = 218.206 g/mol
0.320 g cinnamylidenemalonic acid = 0.0014665 mol
assuming ideal 58% practical yield, theoretical yield = (0.0014665 mol * 100) / 58 = 0.0025285 mol
this corresponds to a malonic acid content of 0.0025285 mol * 104.061 g/mol = 0.263 g
and thus a Ca malonate.2H2O content of 0.0025285 mol * 178.149 g/mol = 0.450 g
0.450 g * 100 / 1.5 g = 30%
so that means that in the best case scenario, the precipitated calcium salts obtained from the ammoniacal H2O2
oxidation of malic acid contains 30% calcium malonate dihydrate. Although judging from my own results, more realistically, I would estimate the real
number to be closer to 10-15%.
For what the rest of the solids consist of, an educated guess would be calcium glyoxylate and calcium oxalate perhaps, both are insoluble in water and
both are likely to be expected as oxidation products of malic acid. I would bet that their calcium content would probably be close to Subramaniam's
21.7%
Attachment: cinnamylidenemalonic acid_hoogen2000.pdf (189kB)
This file has been downloaded 21 times
Attachment: malic acid oxidation using ammoniacal H2O2_Subramaniam1929.pdf (173kB)
This file has been downloaded 15 times
sic transit gloria mundi
|
|
|
| Pages:
1
..
6
7
8
9 |









